Hello,
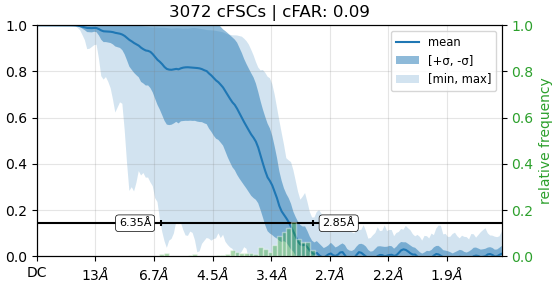
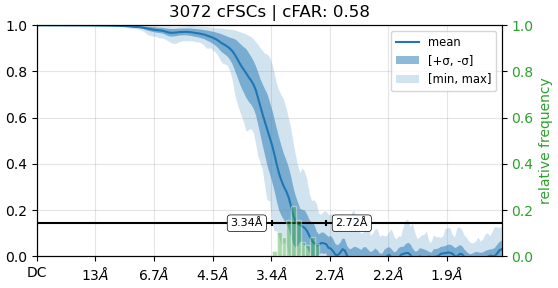
I refined my map using C2 symmetry (after homogeneous refinement), which resulted in a resolution of 3.06 Å and a cFAR of 0.58. However, when I performed the refinement using C1 symmetry (with the same set of particles), the resolution dropped to 3.15 Å and the cFAR decreased significantly to 0.09. The only parameter I changed was the symmetry.
I would like to understand whether symmetry plays a significant role in this case, and how to determine the actual symmetry of the map.
Additionally, although I obtained the map, the models available in the PDB or AlphaFold do not fit properly. Is there any tool or method that can help with fitting the molecule into the map?
Thank you.
Hello,
Just from the look of these two FSC curves, it seems like C2 symmetry is beneficial. Not so much to improve resolution, as the difference between 3.06 and 3.15 is marginal. But symmetry definitely seems to compensate for some preferred orientation in this case (since it gives a narrower range of directional resolutions and a more favorable cFAR).
Regarding your question about fitting an atomic model, it is difficult to help with so little information. There are many ways in which an atomic model may not fit into a map properly.
But if your map happens to have the wrong chirality (this happens with a 50% probability when doing 3D reconstruction from 2D projections), then of course you won’t manage to fit an atomic model at all. Try finding well-resolved alpha helices in your map, and determine if they are left- or right-handed helices. This will tell you if the map has the correct chirality (alpha helices should be right-handed). If the chirality is wrong, you can generate the mirror image of your map with the Volume tools job in CryoSPARC (use the parameter “Flip hand”).
I hope this helps!
4 Likes
Thank you for your response. I will check with the flip map.