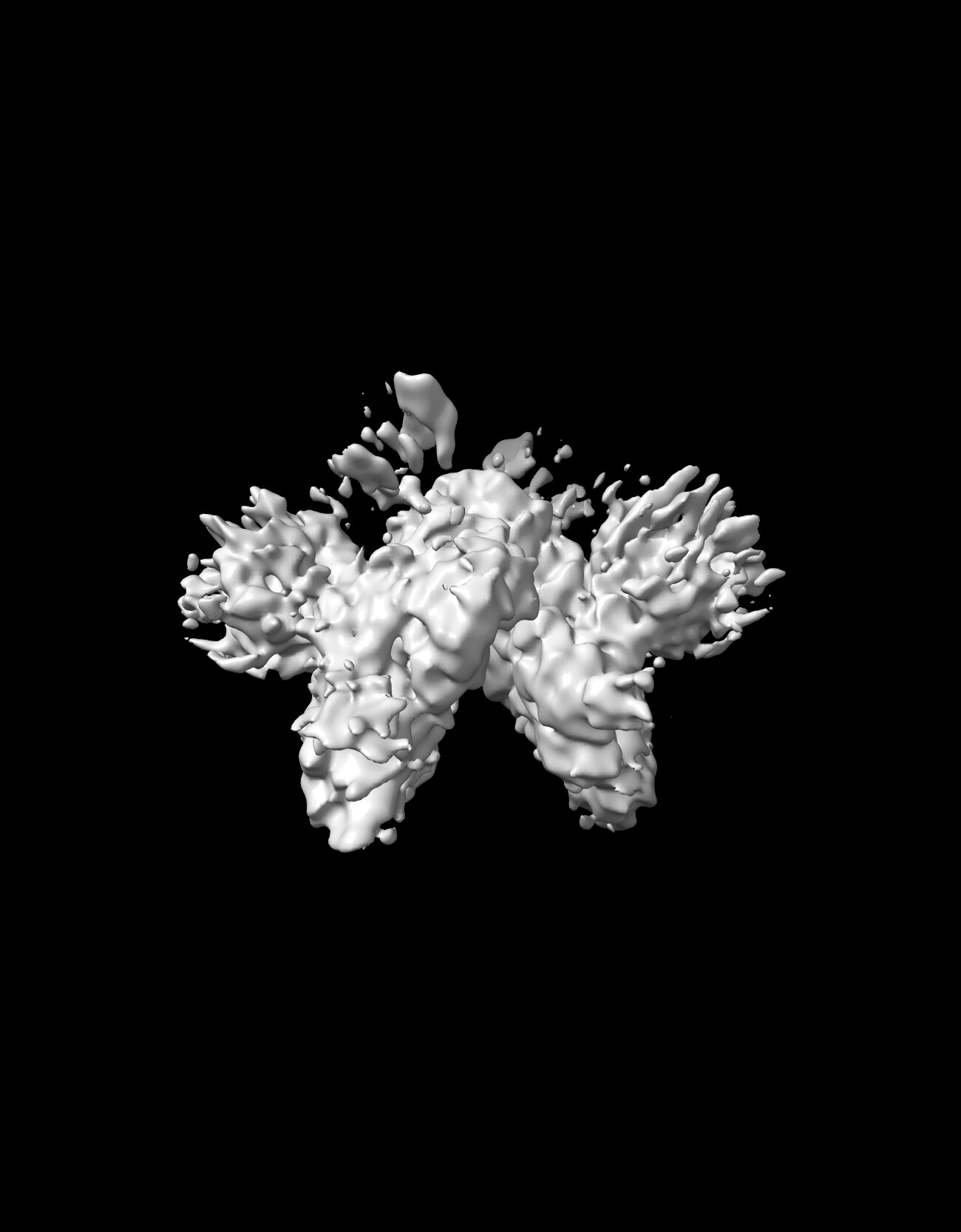
I am new to the structural world but taken some steps toward solving my first CryoEM structure. My protein is ~170KDa. It is a disulphide linked Dimer that exhibit C2 symmetry. The core of the protein is rigid, however the two domains at the periphery are moving independent of each other. I have been trying and experimenting with masks, 3DVA, and C1 vs C2 symmetry in CS and some 3D focused classification on relion but not getting there yet. Any thoughts about how to get the full details of those periphery domains. In the attached image, you can see i might be getting a glance at what could be there or a cloud that lack details
Hi Bassem, the map looks very promising and like you are on the right track. See the discussion here for some recommendations on how to assess pseudosymmetry in cryosparc.
At first glance, I would try symmetry expansion, followed by local refinement with a mask on the peripheral domain (or single protomer), followed by 3D classification without alignments/shifts in relion using a similar peripheral domain mask. Does 3DVA demonstrate large motions on the periphery and inform on where to place a mask and fulcrum for local refinement?
Hi @user123, all those strategies did not seems to work. I believe owing to the size of the molecule. i am trying now to collect particles de novo in relion. Not sure yet how that will pan out.
Symmetry relaxation in Relion was useful to get better results for pseudo symmetry for one of the reconstruction and symmetry expansion + 3d classification without alignment for another. However, your reconstructions should be in correct symmetry axis otherwise it wont work. Basically when you open the map on chimera you should be looking through the sym axis. We requested cryosparc to implement symmetry relaxation, it may be helpful to endorse it so cryosparc scientist may prioritize it.
You need to align your reference at C2 symmetry axis. Then do a refinement on Relion with C2 (you can also try C1) using usal global alignment settings. Then you need run a local refinement with C1 and use relax symmetry option C2 with global and local search angles to be set to same value like 1.8 (that is how you relion runs a local refinement). You need one of the newer versions of Relion3.1 or Relion4.
My group introduced the symmetry relaxation feature to RELION. I agree, it would be great to have a similar option in CryoSPARC! We believe that this feature is very useful when a complex has pseudo symmetry. Note that relaxing symmetry is different from “symmetry expansion + focussed classification” approach. Both have their own use cases. I would be happy to discuss this further with the cryoSPARC team.
Thank you @Juha for developing that, which was instrumental for our mGlu2 structural studies. The importance of this tool has been better appreciated after seeing similar challenges in analogous Class C GPCR reconstructions published lately.
You can remove dust using Chimera but I would not suggest it. This looks fine to me and dust is not harmful in most cases. Just make sure to use low pass filtering for your next refinement step. You can also look at the maps in previous iterations. They may have less dust.
I have a similar situation of pseudosymmetry in a dataset I am analyzing right now. I am super new to relion and was wondering if the symmetry relaxation was done via command line or in relion gui? I am unable to find the option in the gui.
If it’s command line then could you please direct me where I could find the syntax?
Hi @chari1, it is an option in the GUI (at least in 3.1, I assume it is also there in 4). It is an option in 3D-autorefine, I think under sampling. Basically you want to first refine with the “approximate” symmetry in either relion or cryosparc, then refine in Relion in C1 starting from local searches, with symmetry relaxation set to the approximate symmetry of your molecule. Does that make sense?
Running local refinement in Relion is a bit hidden in Relion. In order to run local refinement you need to set Initial angular sampling and local searches from auto-sampling values same (example: 1.8 degrees). As Oliver said you need to first run your refinement with the overall symmetry. You can also run it without symmetry first but your reconstruction should be done where symmetry axes aligned to z-axis, which is also a required step for a refinement with symmetry. Basically the symmetry axis of your initial reference map should be aligned to z-axis. In order to check that, you can open your map with a new chimera(x) session. If the map initially open in an orientation where you are looking through the symmetry axis, then your initial reference map is properly oriented.
By the way currently, only C type symmetry works with Relion symmetry relaxation like C2,C3, etc according to Takanori. D type symmetry relaxation is not working with the official Relion release. You can check ccpem emails for details. If you have non-Cx symmetry you can use symmetry expansion/classification without alignment route.
@alburse have you experimented at all with combining symmetry expansion and symmetry relaxation? For C2 it wouldn’t work, but for higher Cn symmetries it would seem one could safely symmetry expand and refine with symmetry relaxation, so long as a given particle copy is never superimposed on its original orientation (only on the n-1 other orientations) - not quite sure if there is a way to avoid that though…
I need a quick clarification with the parameter settings in the 3D auto refine job in Relion. For symmetry relaxation, I basically provide my symmetry refined map as the input reference map?
Then refine in C1.
Do I need to provide a mask?
What values do I set for:
Ref map is on absolute grey scale - Under Reference tab
@chari1 Sounds like you got it. “Relaxation” just means that each particle will always have a chance to be aligned with some weight to each of the symmetry-related poses.
So basically:
Refine in Cn first
Refine/classify in C1 with local search & relaxation(--sym C1 --relax_sym Cn, same value for “Initial sampling” and “local sampling from”, initial lowpass should be your refine res + 3 A)
More notes:
You can use a whole particle mask, or a focused mask, but it should include each symmetry related unit. E.g. for SARS-CoV-2 Spike you could use a mask for the whole particle, or one that only included the three RBDs. In this case, I would even make a combo mask that included the “up” or “down” state for each RBD superimposed. But all three subunits should be included, it’s not the same as symmetry expansion.
Breaking the symmetry of the reference (e.g. erasing all but one RBD) may help.
Ref. map absolute scale should always be No (for maps made by Relion itself, you can say yes, but there is no need).
Mask diameter should be the diameter of the particle + 30 A or so (to account for CTF delocalization). This is the same as what cryoSPARC is doing with that 0.85 value at the top of every job type. You can use box size in Angstrom * 0.85 just like cryoSPARC. Smaller values might help if the particle is a lot smaller than the box or it’s very crowded.
@DanielAsarnow I was working on the relaxation scheme with different parameters. One quick question, my protein is all in all 160Kda, and the moving domains breaking dimer feature is in the ~30KDa range or 60 since it is on both sides. My 2D classes show them a detail-less clouds. Do you think masking would still be beneficial there?
@olibclarke
Symmetry expansion plus symmetry relaxation could actually be a good workaround for higher Cn but I have not tried the combination since my cases were generally C2. However, I tried either relaxation or expansion and one or the other can give a superior result even in similar cases. Like always, we have to try everything to reach the best possible outcome even though I thought sym relaxation should have been superior to expansion strategy.
@Bassem people often ask questions like this - “do you think X might work given these caveats?” My answer is almost always that I don’t know, but that trying will only cost you a few hours of GPU time. Just go for it!
Of course, I have lots of opinions about what parameters to use, like in the post above.