I’m using homogeneous and non uniform refinement at the moment on a pseudosymmetric assembly, and I’m wondering how the branch and bound algorithm used for these jobs copes with cases where there are multiple poses that are almost equivalent in terms of score, but very different in orientation.
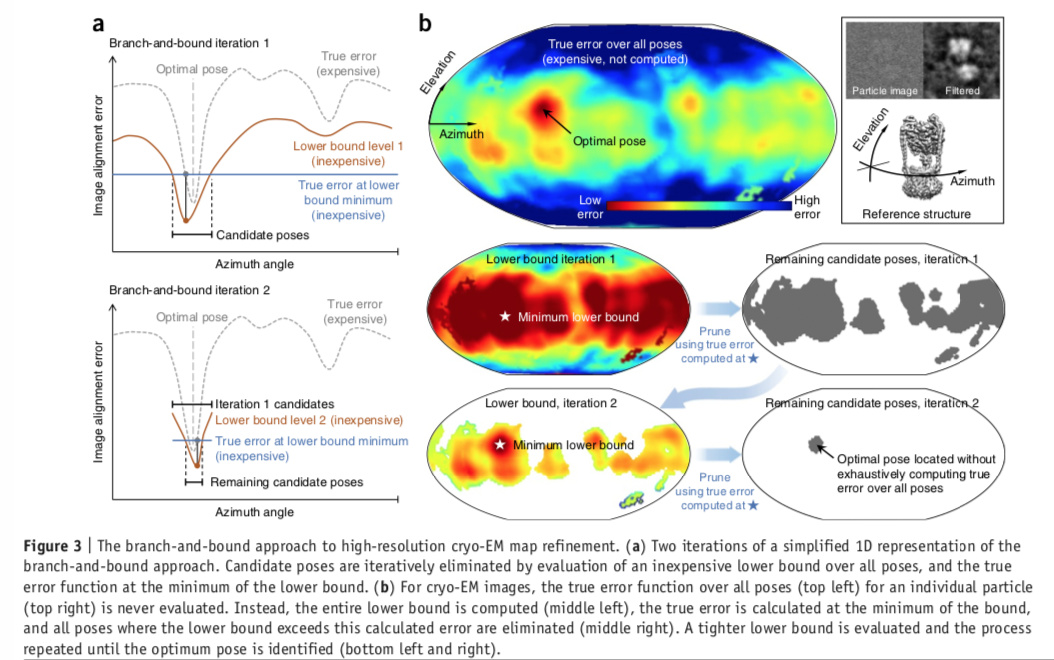
Looking at Fig 3 of the original cryosparc paper, it seems like there is a possibility that for some particles the correct pose could be eliminated if the minima corresponding to the pseudosymmetrically related poses are close together in terms of score. Are there any parameters one can tweak to control how aggressively cryosparc prunes the search space at each iteration? So that one can search more comprehensively, at the expense of speed?
My understanding is if there’s another minimum that juts down below the blue line in iteration 1 (top of part A), that part of the space is retained in iteration 2. It should keep the pseudosymmetric false minima across the iterations until the bound excludes them. Restated, eventually any non-marginalizing reconstruction will have to choose one alignment parameter; in cryoSPARC this should happen only after projection-matching against the current reconstruction gives error estimates that place the alternate parameters above the lower bound.
In a marginalizing program like Relion, you could instead end up with significant alignments at both minima and then have each particle inserted e.g. 40% at one and 60% at the other. Personally, I would rather have the single correct answer, but this approach can work well for some data (as we have no doubt all seen when using Relion).
The final option is something like pseudosymmetric refinement in Frealign (and the forthcoming cisTEM 2). There, the symmetry group is given with a lower-case code (“c4” instead of “C4”). Then the metadata file gets a line for each particle at each possible symmetry-related pose, each with its own occupancy and score. These scores are used for per-particle b-factor weighting, so particles can contribute low-frequency information to all symmetry-related poses and more high-frequency information just to the higher scoring one.
Assuming my understanding about cryoSPARC above is correct, then a possible tweak would be to always retain the poses which are symmetry-related to the best pose when computing the true error. This could go along well with limiting the search space for symmetric refinements, which cryoSPARC hasn’t done previously. It could then be requested using a checkbox for pseudosymmetric refinement, similar to giving a lower-case point group in Frealign.
@DanielAsarnow is correct that pseudosymmetry-related poses (that have similar residual error) should be retained until the latest iterations of BnB, especially since in cryoSPARC we don’t limit search to the asymmetric unit. But in practice due to the way poses are discretized for branching/bounding (and the interplay with discretized shifts), the pseudosymmetry-related poses may never be exactly compared and the correct one may get overlooked towards the end of BnB. e.g. consider a symmetry where the angle between asymmetrix units is not 90 degrees - this means already that the symmetry cannot be exactly represented on a cartesian grid.
I think marginalization (e.g. Relion) has a similar issue that the true pose will only end up with a larger weight if the fine-ness of sampling poses is high enough that a direct comparison is made between the pseudosymmetrically related poses. I’m not sure at what sampling level this would be fulfilled. The best approaches are definitely those that are specifically designed for pseudosymmetry. The suggestion @DanielAsarnow made for how to do this in cryoSPARC I think is the right direction. It’s on our list now
@olibclarke@apunjani thank you for bringing this important topic up. If you have a protein complex with 4 subunits, 3 of which are indentical, and the 4th is different from the other three by a disordered region which is not visible in the map, would it make sense to impose C4 symmetry or what artefacts could it bring ?
On a related subject, we recently noticed that the template picker or blob picker some times produce picking results that is off-center when the protein is small and the particles are quite crowded in the micrographs. Our current strategy relies on using 2D classification and heterorefinement to find our target particles, but we notice that quite a large number of particles get thrown away during hetero refinement. After throwing these ‘bad’ particles into a local-refinement job, we find that quite a large number of the excluded particles are not actually bad particles, they are just off-center and maybe hetero-refinement is failing to find such off-center optimal pose. I also notice that no alignment input slot is required for the hetero refine job type, so I’m wondering if the translation searching grid is always centered on the image center (the picked particle center by blob/template picker) and aligning the particles to a 3D reference with a large search range using local-refinement might help in this case.
Our current work around is to get a reasonable 3D reference first. We would then return to the initial dataset and use local refinement with large search ranges to center the particles and perform re-extraction using the updated alignment parameters. This approach has been proven to be useful for some of the cases, tripling the effective number of particles, but poses additional disk usage.
I wonder if there is a way to tweak the bnb parameters and increase the search range as well as the tolerance to alignment error lower bound for the poses kept at each bnb iteration. Also, I’m still a little naive about the way cryosparc generates the shifting grid to search over. The Supplementary note 2 in 2017 Nature methods paper mentions that the first bnb iteration uses a spacing of 5 pixels for translation search, does the first bnb search iteration loop over the entire search grid, or is the search limited to a certain number of pixels around the grid center? If so, is there a way to modify the search range and search step of the bnb iterations ?
It sounds like you may be able to make your picking more accurate, by tuning the minimum interparticle distance, low-pass resolution of template and micrograph, and the number of maxima to consider. Use runs on 10 micrographs only, to iterate rapidly.
Say your smaller view is 100 Å and the larger maybe 160 Å, then perhaps try using a low pass resolution of 15 Å, a minimum distance of 0.2 - 0.4 the (max) particle diameter, trying the elliptical and circular blobs at the same time, and tuning maxima from 2000 - 5000 in steps of 500 or 1000.
I use this strategy to get good picks, including on samples that are considered “difficult,” just through several rounds of trial-and-error. The goal should be to pick well, but also over-pick, and then use the inspect picks job to throw away only ice and obvious background pics. I am generous at this stage, the thresholds are not the same on all micrographs due to defocus changes and edges, so you can err on wide thresholds.
PS An easy way to recognize that further tuning is needed is if the raw picks look like they are scattered randomly and then “repelled” each other into a more or less uniform spacing (due to minimum interparticle distance).