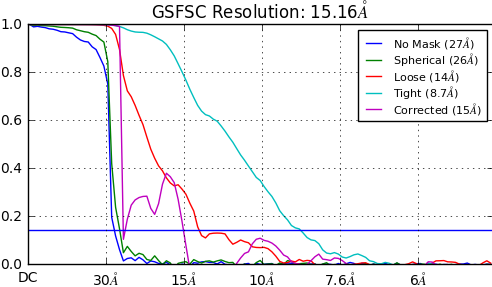
I am getting artefactual FSCs when running the homo_refine programme. It is the artefact common when masking too tightly i.e. the corrected FSC drops to zero at the resolution of phase randomisation.
I tried supplying my own refinement mask rather than the dynamic one made by the programme and the problem persists. It appears that the mask used to calculate the FSC is different to the user provided one (…mask_refine.mrc vs …mask_fsc.mrc).
Would it be possible to build in a feature to use a user provided mask for fsc calculation or an advanced parameter to tweak the tightness of the mask made to calculate the fsc?
This artefact is limiting the resolution of my refinement, as it lowers the resolution of the data used for the next round of refinement.
I would really appreciate an answer to this question, as this is preventing me using this software for a particular project.
I have examined the masking in the refinement. The mask_fsc file appears to be excluding portions of my protein, therefore setting the contour threshold for the mask incorrectly.
I have tried fiddling with the masking options available in the refinement job, however I think this only changes the refinement rather than the FSC mask.
Could you please confirm whether the masking as part of the FSC calculation is dependent on these parameters and whether there is a user intervention possible to change the mask used for the FSC?
I have attached an example FSC below as a demonstration of the problem.
You can use a static mask as you would in Relion. There are situations where no “adaptive” (percentile-based) method will lead to a correct result - in such cases you will always need to create a mask manually by volume editing in Chimera.
Sorry for the delay in replying to this point, thanks for reporting.
Have you already found a way to address the problem? (perhaps new/more data?)
Currently in cryoSPARC the FSC mask is auto-generated separately from the refinement mask, to ensure that a user does not input a mask that would cause masking bias in the FSC. Of course in your case this is backfiring… but looking at the FSC curves, it’s concerning that even in the nomask and spherical mask cases, there is no correlation beyond 30A. Generally speaking, if the data is good and alignments are good, even with a 30A reference in one iteration, the next iteration should show strong correlation well beyond 30A with the spherical mask.
Thanks for the response. I have not really found a way to solve this. The issue with this particular project is that there is an amount of disordered density at the surface of my protein, which I think doesn’t work well with your masking algorithms.
The lack of correlation at higher resolution is owing to the fact that this is a relatively elongated particle in a large box. The ‘tight’ mask resolution from these refinements is reasonable if I recalculate FSCs from the half maps.
This is a slightly special case, as I have not seen this problem with lots of other datasets.
I think a really useful feature for me would be to be able to use the option to use the tight mask resolution for refinement (with caution) and do the correction manually afterwards or an option to use a user defined mask for FSC calculation.