Hi everyone—I’m working on a project to determine the structure of a ~50kDa membrane protein with a transmembrane region and an extracellular domain. After 2D classification, ab initio reconstruction, and 3D refinement, the TM region shows density but no discernible helices. In this situation, should I (i) build a TM mask from the refinement-derived volume and run another round of refinement, or (ii) generate a mask in ChimeraX based on the AlphaFold prediction and use that instead? I’m also unsure how best to proceed once I have a TM mask when helices aren’t visible—would it be better to reclassify to obtain a cleaner particle set first?
50 kDa is pretty small for starting point, is this monomeric? How many transmembrane helices? Is the transmembrane connection to extracellular domain expected to be structured, is extracellular visible? Is there room in the current TM density for the TM helices predicted? If expecting structured between extracellular and TM then the lack of any TM helices density is troubling; cleaning up this particle stack likely better, but would still try local refine on TM region and 3d classification/3DVA on the locally refined maps. How many particles? If not structured between TM and extracellular you’ll want a large number (500,000+, ideally millions) as micelle tends to dominate vs few TM helices.
Four transmembrane helices are about ~10-15 kDa, i’ve seen positive results for a complex (100’s kDa) that interacts with an ~50 kDa protein with 4 transmembrane domains. There was initial rough estimates for this 50 kDa protein for where its 4 transmembrane helices were interacting to the complex in non-uniform refinement that upon local refinement was able to get some side chains out of the transmembrane region. But this was nicely structured protein between transmembrane and extracellular in addition to having the large complex as a good starting point.
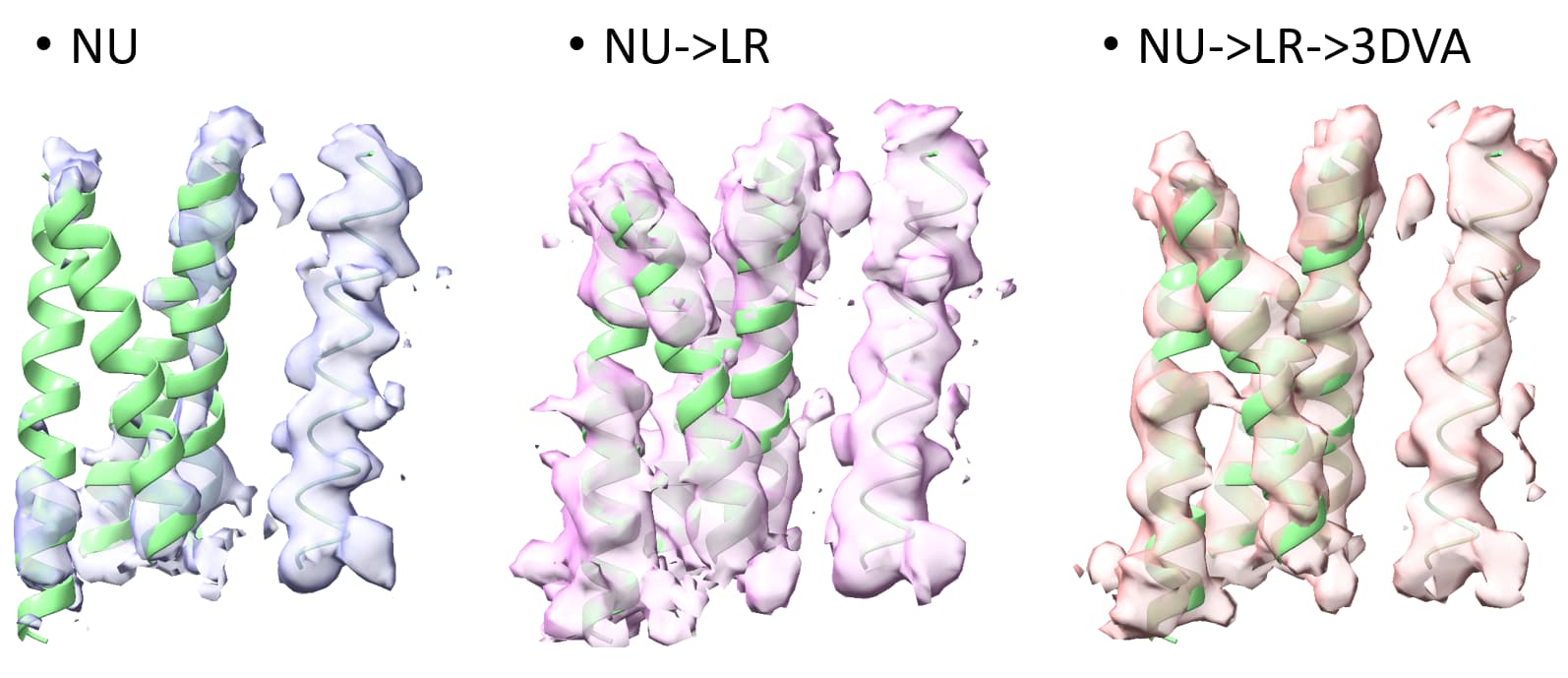
Above image the 4TMs of small (50kDa) interactor getting better with a single TM helix of rest of complex on right.
Not all membrane proteins have nicely resolvable transmembrane domains depending on the conditions (detergent?). I’ve had cases where i know the protein, based on mass spec and analogs, should have TM domains but unable to see any hint of some of the expected transmembrane helices even though the extracellular domain goes to sub 3A and some visible TMs sub 3.5A. Local refinement improves those transmembrane domains that are visible but was not able to observe the missing TM helices upon LR or any classification (heterogenous/3Dclass/3DVA).
Apologies for the delayed response. My protein consists of five transmembrane helices, and the extracellular domain is not rigidly connected to the TM region. After reconstruction, there is density within the detergent micelle, but the five helices remain undistinguishable. The extracellular region is visible, and the overall structure shows around 10 Å resolution after refinement. Currently, I don’t have better strategies for optimization. Someone suggested using AlphaFold to predict the structure and create a mask for subsequent refinement, but as I’m still in the learning phase, I’m not entirely sure how this would aid in structural analysis.(The image below shows my refined structure.)
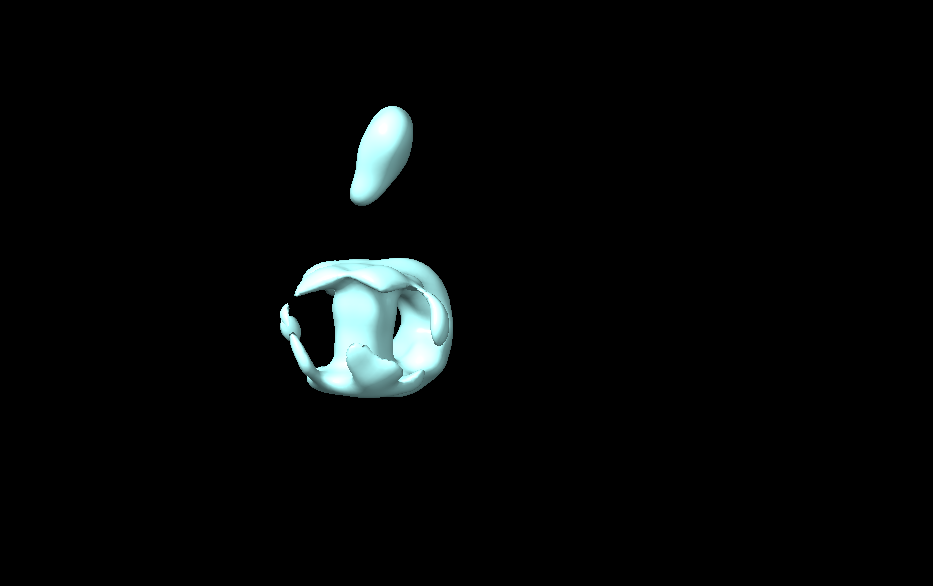
This suggestion is a little dangerous, as it potentially introduces masking bias. I’d not recommend doing this.
…
What do 2D classes look like? Can you see protein (TM helices) clearly in the 2D?
Thank you for your reply. To be honest, I’m not entirely sure how to implement this suggestion either, haha. My 2D structure does show some helices, but they aren’t very clear, as seen in the image below.
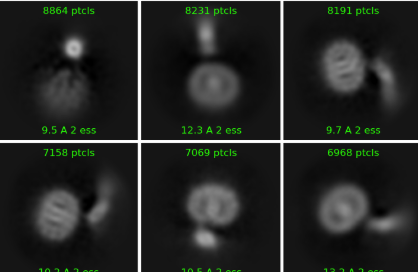
1 Like
I’d say you’ve not got enough particles to get to high resolution for something so small… ![]()
It’s encouraging that you can see distinct helices in 2D at all for something so small!
How big is your total stack (going into 2D)? I would suggest redoing 2D with 80 O-EM iterations, 10 final iterations, 400 batchsize, max resolution for reconstruction 3 Å, using unbinned particles.
For small things like this, sometimes the data in the 6-3Å range can really help for alignment, and it will give you an idea of how realistic high res alignment in 3D is.
2 Likes
Thank you for your reply. This is my second round of 2D results, with approximately 750,000 particles. The first round of 2D had over 2 million particles. For my 2D, I used 50 O-EM iterations, 1 final iteration, a batch size of 400, and a max resolution for reconstruction of 6 Å. I will try to follow your suggestions and run it again.
Yes, that’s why I’m trying, but it feels a bit challenging.
![]()
1 Like