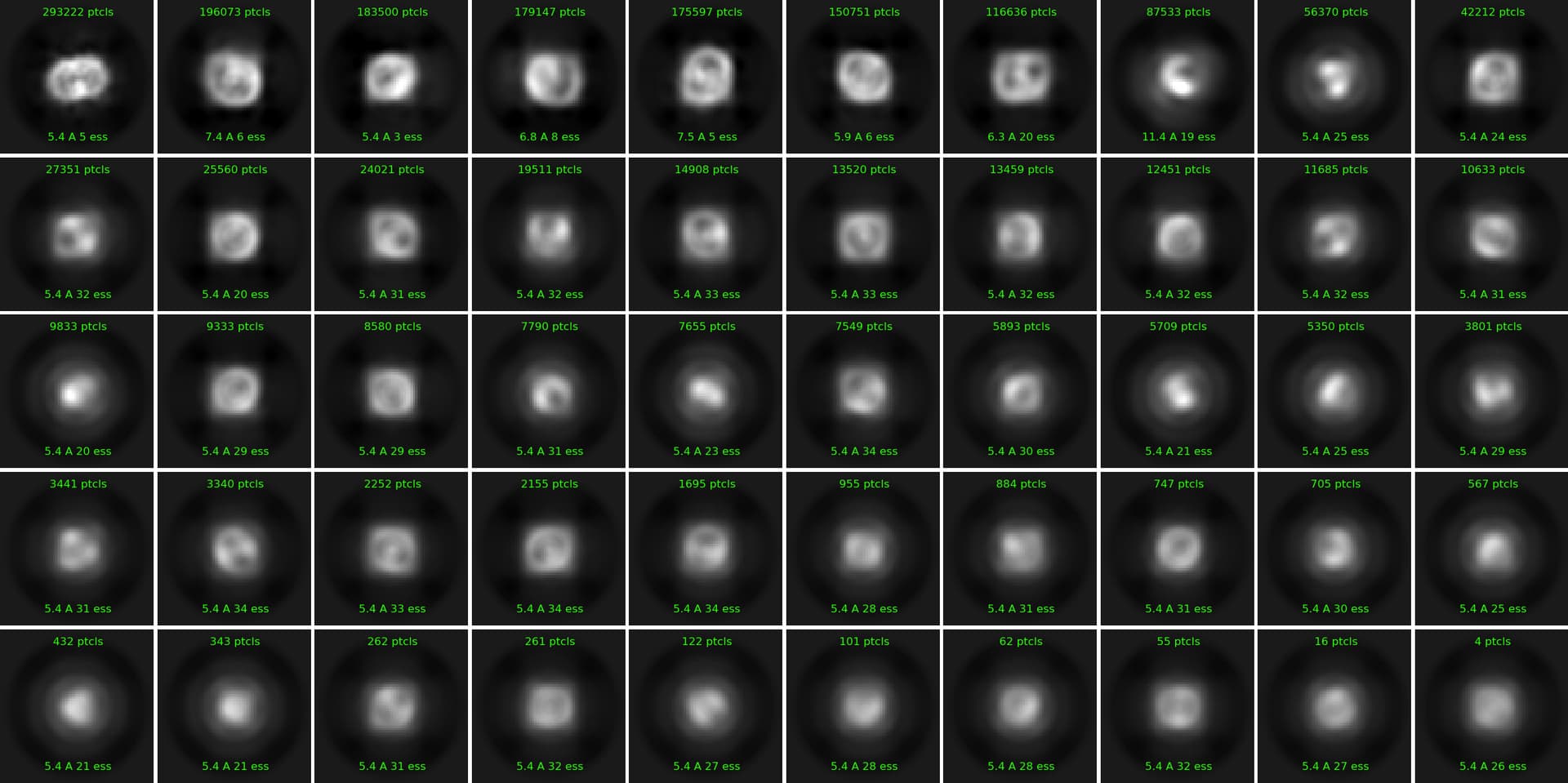
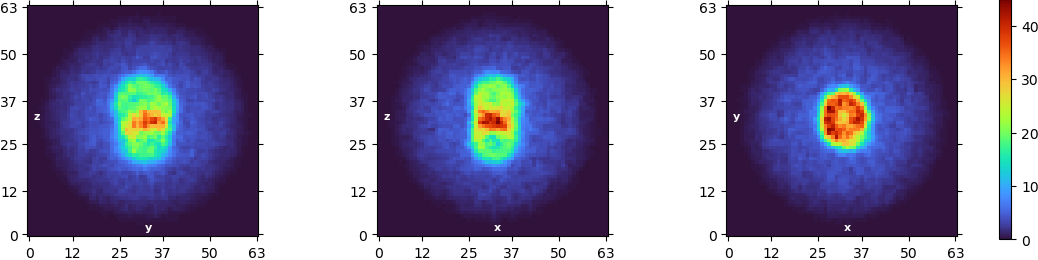
Hey everyone, I am new to cryosparc and processing SPA data the first time. I have a data set of a small membrane protein bound to a hexameric soluble protein. The initial 2D classes are looking like that:
. I used 50 classes with max resolution of 6A and FORCE MAX OVer POSES= OFF. They already look very “hazy” and I cannot really see any helices or other features. I selected the ones that resembled micelles and tried an ab initio. The parameters used were:
number of classes=4
max. resolution=8
initial minibatach size =90
final minibatch size =300.
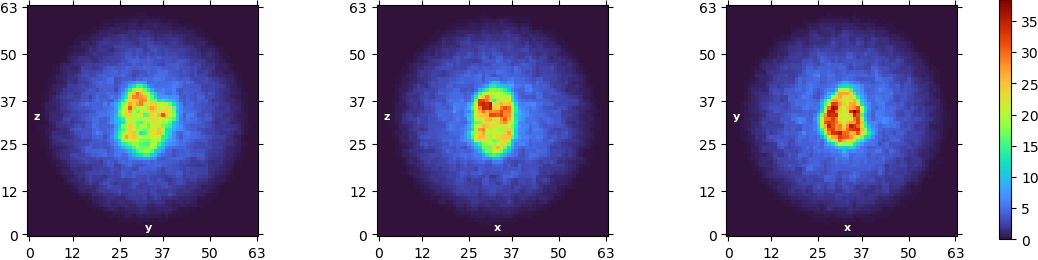
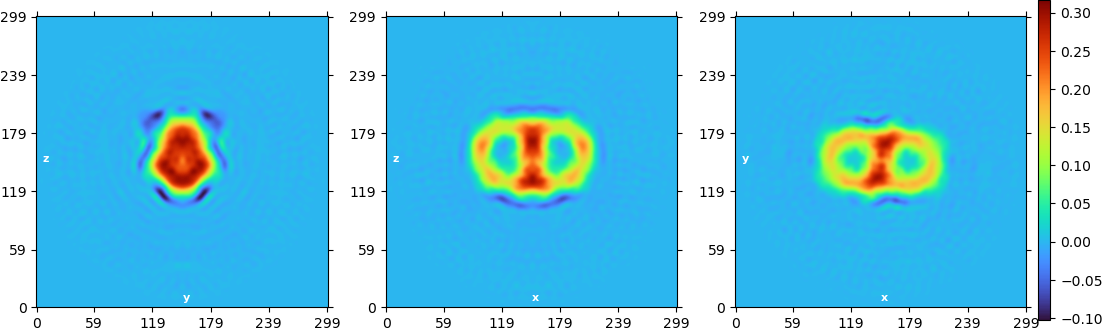
I got the following abinitio reconstructions:
The resolution is now “stuck” at 8A and I cannot really see anything resembling the membrane protein. My question is: are the 2D classes just artefacts or actual detergent micelles? And how can I improve the resolution a bit further. At the moment I am bit lost, with the density I got out.
Any help is appreciated!!
Cheers, Sa
What box size are you using? It looks on the small side. Also how many particles do you have in your dataset. And in your 2D, you may want to increase the number of iterations (to 40 or 60), this does not look like it has converged.
Also, are you 100% sure you have C1 symmetry in your NU refine job? To me it looks like there are symmetry artefacts in these slices.
Hi @olibclarke,
thanks for your fast answer! my box size is 200px for extraction. and I used a box size of 80 to 160A (Blobpicker). In total, there are ~ 1,700 000 particles. I am running 2D with 40 and 60 iterations now. Let see how this will look.
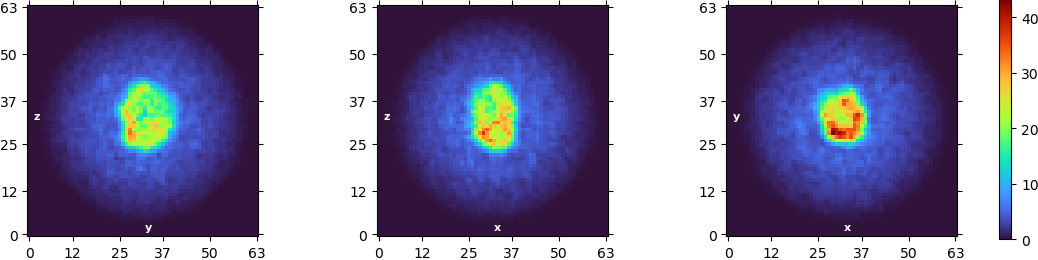
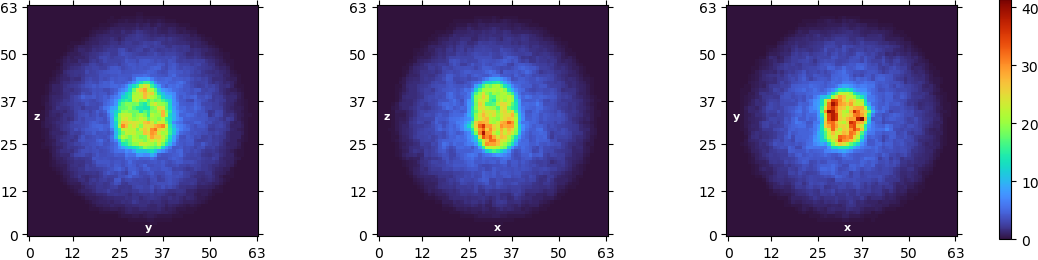
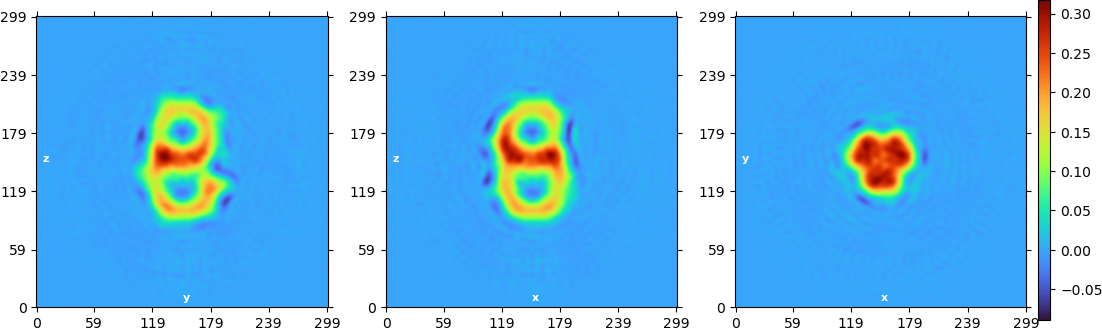
Concerning the symmetry, I had the similar thought and tried NU refinement with a C3 symmetry applied and I got that:
I would try a larger box size - 200px is likely too small. What is your pixel size? Also, with this many particles I would use more classes for your initial 2D - 200 would be a usual starting point.
Also re symmetry, I would initially use C1 symmetry. I would not trust any resolution improvements you see from enforcing symmetry unless accompanied by genuinely improved density (e.g. appearance of secondary structural elements).
Having said that, if you suspect that your protein has symmetry, and C1 ab initio is not converging on anything sensible, it may be worth testing enforcement of different symmetries during ab initio. Sometimes this can allow convergence to a correct solution when C1 will not converge (but again, only trust it if you see “real” protein-like features appearing in the maps).
I don’t think the first images you posted from your NU refine are C1 symmetry - you may want to double check that, as there appear to be symmetric artefacts in all three slices.