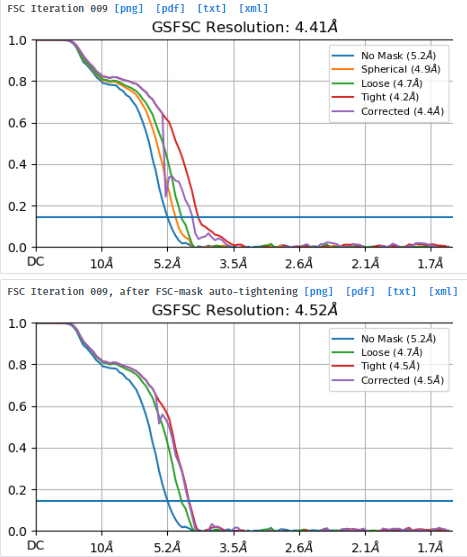
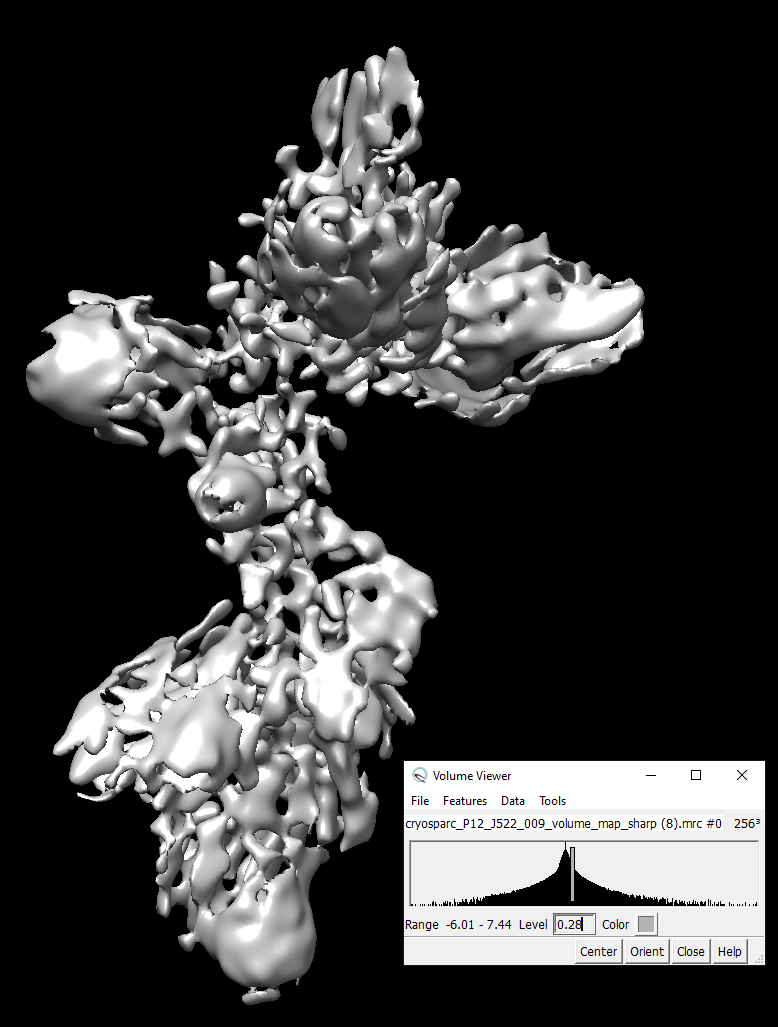
I suppose the RNA helices can be visible in the density map of 4.52Å. However, it can not be identified.
Did cryosparc overestimate the resolution for RNA structure? Or, I need to go further refinement. What should I do?
Huge overfitting - the flower-like spikes are a dead give away. What do 2D classes look like? I would suggest cleaning up your particle set more, and also trying non-uniform refinement.
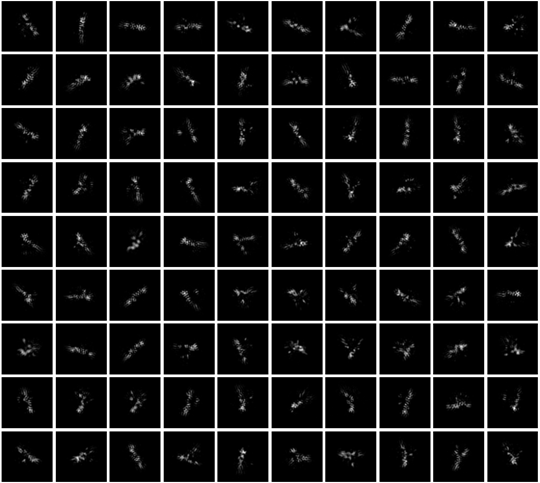
Here is the 2D classification of the 400,000 paticles.
I tried to select a better subset of 200,000 particles and run heterogeneous refinement. It looked similar.
I will try it again and test non-uniform refinement.
I know the flower-like spikes are problems. How do you know the spikes are due to overfitting?
I don’t know that I see any problems with your 2D classes (although I have never seen a 2D class from an RNA structure before! Very cool!). It might be worth keeping the 400k particles and trying to weed out any noise in 3D instead.
I have a lot of heterogeneity in my own data. My favorite starting method is to do a one-class ab initio, then multiple classes of a heterogeneous refinement. Sometimes it works to do a multi-class ab initio first. I discard whatever class/es looks like a noisy class, and continue with the rest of the particles.
Depending on what the classes look like at this point and how similar they are I might combine all or a subset into another ab initio and heterogeneous refinement, or just continue treating each class separately. After the heterogeneous refinement I do a separate NU-refinement on any classes that look promising and like there are enough particles.
If the heterogenous classes are really different I try to pull out different conformations through 3DVA instead. I do an ab initio and homogenous refinement on all the good particles, and use the homogenous refinement output as the mask input for a 3DVA job. Then before running 3DVA I do a NU-refinement on the ab initio output. I combine the NU-refinement particle set with the homogenous refinement mask to feed into 3DVA. In my case it’s taken a LOT of tweaking of 3DVA parameters to find the sweet spot for my dataset.
After 3DVA I do a couple of 3DVA analysis jobs. Sometimes sorting by intermediate states gives me enough to pull out similar particles, and sometimes clustering works better. I combine a certain number of intermediates, or the clusters that seem to be most similar, and do another ab initio and NU-refinement. Again though, the workflow and parameters will be specific to the dataset you’re working with.
I had some of these pesky shell-like “flower spike” densities that I had a hard time getting rid of. They’re related to overfitting within the masked region I believe. One thing that worked for me was waiting to start dynamic masking until a higher resolution. AKA waiting until something like 7A instead of the default which I think is 12A. If you’re doing dynamic masking it might also help to play around with the Dynamic mask near and far parameters.
Here’s a good thread that discussed troubleshooting these spikes as well as some other stuff:
Your 2D classes look very good. How many rounds of 2D have you performed? Did you optimize the band tightness on native gel and is the secondary structure information for this sample predicted or experimental?
I would try doing ab inito with 1 and several classes, and the maximum resolution set a bit higher like 8 Å. Are they consistent with the refinement too? If so how do they look?
For your refinements, @jenchem is right that you should mask more conservatively. I would disable masking entirely by setting the resolution to 1 Å. The 2D classes seem consistent with refinement structure, maybe using no masking and controlling the alignment resolution and/or low-pass filtering the map will help.
Hi @jenchem,
Many thanks for your suggestions.
Due to the structural flexibility and heterogeneity of RNA, I did not expect we could get a high-resolution density map for the 250nt RNA. I think there must be some problems (such as overfitting) and artefacts which should be removed. Thank you for sending me the detailed information. I will follow it to find a good solution.
We ran 3 rounds 2D classification. These classes can only be seen when we enable “Enforce non-negativity” and “Use clamp-solvent to solve 2D classes” for 2D classification. It may due to the low contrast of our RNA samples in images. The secondary structure of the RNA is predicted by computational methods together with Shape-MaP and DMS constraints.
I tried to set a lower resolution for the ab inito. It did not help. I agree I may need to limit the resolution and play around with mask.
My best RNA structures seem to be from letting ab initio go to higher resolutions. I’m surprised the contrast is low, for me it’s strong, but the particles are very heterogeneous.
I got good results using Ab initio with more than 1 class so you don’t discard the good classes in the 2D step. Use a higher resolution range for this. Then 3D classification followed by 3D NU Refinement worked well for me.
@Vruiz that’s interesting, for me I got nothing from ab initio until I did several rounds of 2D classification selecting RNA-like classes. How did you pick originally?
I still make one or two rounds of 2D classification but I only remove clear noise 2D averages. I picked using blob picker and TOPAZ, although topaz made a better job.
Ah, then I think our experience is actually similar. After I used the EMAN2 NN picker (actually a very different than Topaz or BoxNet) and got my first results, I went back and used templates genereated from my 3D volumes to pick.
Did you use phase plates for collecting your RNA data? It seems the phase plate can help to increase RNA sample contrast.
Yes. It’s heterogeneous. But after running multi-class 3D ab initio and heterogeneous refinements, I found some parts of the map look rather consistent. I used a mask to focus on the region for uniform and non-uniform refinement. The FSC looked better. But, still no RNA helices was visible and the flower-like spikes could not be avoided.