Hi everyone,
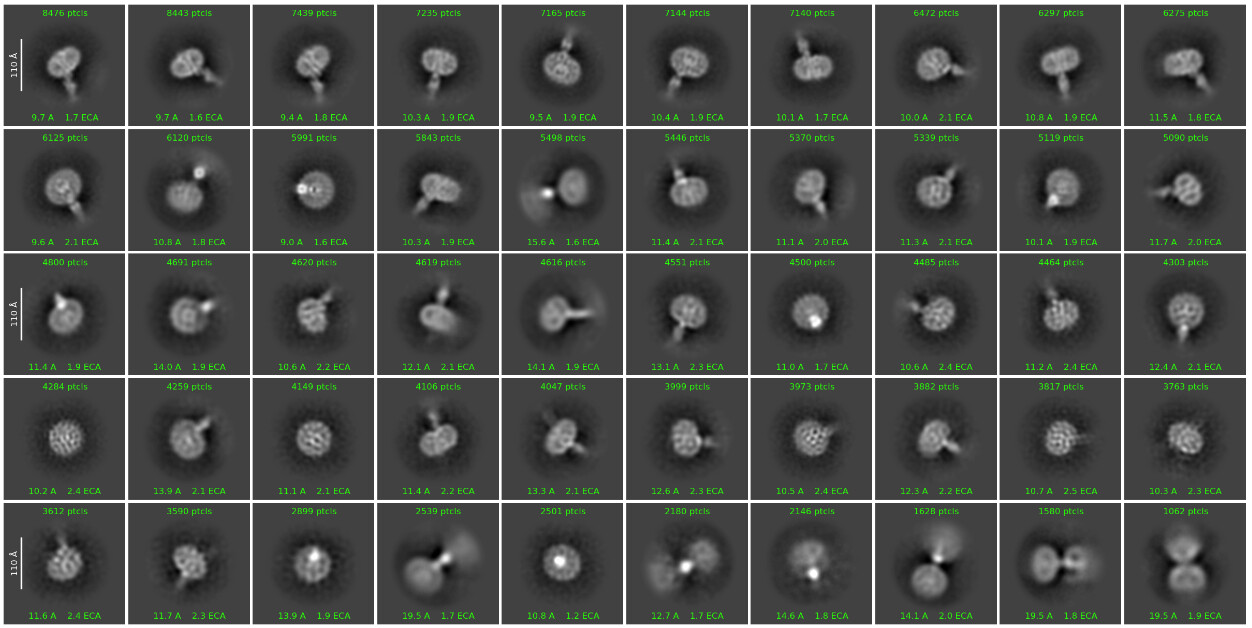
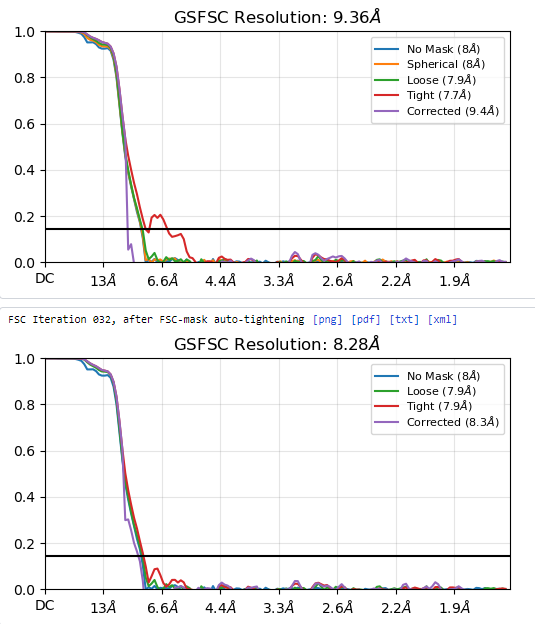
I am currently solving the structure of a complex consisting of a small membrane protein (34 kDa) and a nanobody (13 kDa). Based on the outputs from the 2D classification (Figure 1), Ab-initio (Figure 2), and heterogeneous refinement (Figure 3) jobs, I thought they looked okay. However, the non-uniform refinement (240k particles) job didn’t provide a good map, and its corrected FSC curve dropped at the low-frequency region(Figure 4), which seems very unusual to me.
Figure 1
Figure 2
Figure 3
Figure 4
I’m unsure of what to do next and would appreciate any ideas or suggestions you might have.
Thanks,
Ming
I cannot give you a certain answer, but my 2 cents would be to still clean up the dataset further at the 3D level with heterogenous refinements and multi class ab-initio.
May I ask how much you low pass filter the initial model? Maybe it is worth trying less, like 12-7A.
In my experience, refinement does not improve maps that much (does not take a map from 8-9A below 7) for small membrane proteins. Only if the ab-initio reaches high resolution, will refinement improve on it. Like from 5-6A to below 4A.
Maybe what I can also recommend is to do rather more iterations of heterogenous refinements (like 20-30 final iterations) and use multiple bad classes, that look rather similar to your good class. And for multi class abinitio, you could also use a large number for initial and final iterations.
Hi @MLiziczai, Thank you for your quick reply.
I have questions for your commments.
1.May I ask how much you low pass filter the initial model? Maybe it is worth trying less, like 12-7A.
Do you mean the initial resolution I used for heterogenous refinement? I did use the 8-10 A and it really help.
2.In my experience, refinement does not improve maps that much (does not take a map from 8-9A below 7) for small membrane proteins. Only if the ab-initio reaches high resolution, will refinement improve on it. Like from 5-6A to below 4A.
Could you tell me which job the map below 7 Å is from? In addition, how do you know the resolution of the map from the ab-initio job?
Apart from them, I used downsampled particles (bin4, Nyquist resolution is 7 A) to perform the ab-initio and hetergenous refinement jobs? do you think this will affect the result? If so, should I downsample the particle or not?
.
Thanks,
Ming
Thank you for the great ideas. I still have several questions.
What do you mean by saying the bad classes look rather similar to the good class? Do you mean the ab-initio model from bad class looks like the one from the good class? If so, how do you determine which one is bad class and which one is the good class? Is it based on the resolution you mentioned above? If so, where I can find the resolution of the map from ab-initio job?
Thanks,
Ming
I hope it helps. I do want to stress that I am by no means an expert, so take everything that I say with a grain of salt. Maybe someone who knows more could comment corrections, but this is what I think/experienced:
-
Low pass filtering: for everything, also for ab-initio. I worked on a 52 kDa membrane protein, and setting the initial resolution to 12-7A helped a lot (and final resolution to 7-4A respect.)! You can set it similarly low for all jobs, and once you have a great structure you can test with an ab-initio, where you low pass filter more to 15-20A, to see how biased your map is.
-
Knowing the resolution from ab-initio: you can only guess it. Typically you see solid a-helices as continuous bananas at 7A, and beta-sheets around 5A. If your helices start to show a spiral, you are approaching 4A. If your helices are broken and segmented, you dont really have 7A.
-
Binning and Nyquist at 7A: it has no bad effect, in that case your max res will be 7A, and you would have to unbin for refinement, which I can see that you did. I like to keep it as binned as possible (till I reach Nyquist), just to save space and time.
-
Bad classes and good classes: bad classes should look similar in size and shape. If you have aggregated protein, or in thick ice that cannot contribute to high resolution reconstruction, or empty micelles, and you run the heterogeneous refinement using your good ab-initio model and a bad once that is 1/10th the size (just some random ice particles), the algorithm will sort your unwanted particles to the shape that looks most similar at the given low pass filtering, so something that has approximately the correct size and shape. Then you can use het. ref. where particles that do not improve the resolution of your good class are then sorted to the 3 bad classes (junk trap).
-
Determining good class from bad: well, good one is something where you see your structural features like a-helices, nanobody, etc. Bad ones are the ones where you don’t see these features. If you cannot get one from ab-initio, you might want to generate one by poorly refining your map, that will not look like your protein of interest, but has a similar size and shape.
Marton
Thank you very much, Marton. I will try your great suggestions.
Hi @team,
Could you please do me a favor? I recently attended a lecture by Dr. Jiang, a brilliant expert in cryo-EM data processing. He presented an excellent strategy for achieving high resolution in small membrane proteins. The method involves using a composite mask, which utilizes two masks during 3D classifications and 3D auto-refinements: the regular reference mask that covers only the protein, and a second mask that covers the detergent micelle. The second mask helps reduce noise in the covered region using a low-pass filter. By employing this composite masking technique, the low-resolution information from the detergent micelle constrains the global alignment, while the signals from the protein enable more accurate particle alignment and 3D classification.
However, this composite mask-making approach is currently only available in RELION (available on GitHub: https://github.com/jiangjiansen/relion_composite_masks). I’m wondering if it can be applied in the CryoSPARC software as well. If so, it would be very convenient for CryoSPARC users.
I am looking forward to your response.
Thank you very much,
Jianming
1 Like
Hi Jianming, I can’t speak to potential availability in cryosparc, but a similar strategy is also available in CisTEM. In cryoSPARC, NU regularization should achieve a similar effect, but of course different approaches can work better or worse for different problems.
I guess in CS you could technically take a similar approach in two stages - first running an NU-refine with a limited maximum alignment resolution, to coarse-align on the micelle, and then following up with a local refinement with a mask that excludes the micelle, and a higher maximum alignment resolution. I haven’t tried this though, so not sure how well it works.