Hi everyone!
Today I would like to ask a question on your strategy involving the use of 3D Classification for peeling may conformations from a dataset of a wildly active protein.
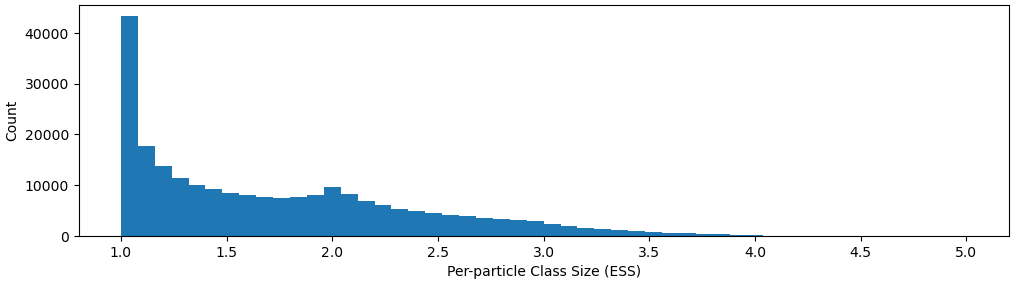
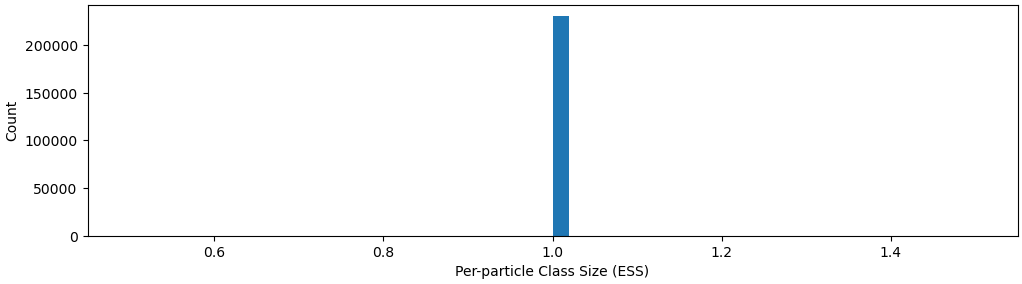
I have been using 3D Classification on 800k particles (selected by many rounds of 2D classification to remove junk particles) and split into 4 classes consisting of 180k, 250k, 198k, 203k particles each. Then, I realize that each class can be split into 2-3 more classes. For one of those subclasses consisting of 40k particles, when I tried 3D Classification again into 2 classes, I got back mostly 39k particles class and another class of junk particles. This I would consider as “exhaustive classification”, where I cannot split 1 class further.
The reason I have been more actively using 3D Classification is because previously we ran the 3D Classification job on our old workstation and it took more than 1 week to finish. Then when we migrated to a faster workstation, we were able to run the job in 2 days, and smaller set of particles in less than 2 hours. Therefore, although I have been building models on the maps I got from the initial 4-class Classification job, I have been actively doing more 3D Classification and discover smaller and more intricate movements of my protein, that got averaged (and lower quality) in the first 4 maps.
My question for our community is, if it is better to do 3D Classification until exhaustive (cannot split anymore, or split into homogenous classes, confirm by NU-Refine and look at the map closely), or is there a better way to classify the highly dynamic states of the protein that got captured in CryoEM? I tried 3D-Variability but that has not really helped me separate the classes.
Moreover, should I be doing “double confirm Classification” where I run the 3D class job again with similar parameters to see if I get the same results? I found one way to ensure the 3D classification job get easily reproducible results is by decreasing the Convergence criterion (%) to much lower numbers, like 0.001, and increase the max F-EM rounds (up to 50-100) to get a very stable classes without much particles shifting between each round of classification. But this would only work on very fast workstations.
Looking forward to hearing your thoughts!