Hi all,
I tried to get some good 2d classfication from around 12K moives. However, the result doesn’t look reliable, seems a bit blurry. I also attached the raw micrograph here. Any suggestion on that?
Hi all,
I tried to get some good 2d classfication from around 12K moives. However, the result doesn’t look reliable, seems a bit blurry. I also attached the raw micrograph here. Any suggestion on that?
You could try setting the circlular mask inner diameter to a value closer to the width of your particle to rescue those picks that have two adjacent particles. That could help you get a better idea of whether that is your protein or contaminating micelles. It also seems like there are a lot of bad particles, so you may be able to clean things up by doing subsequent rounds of 2d classification after eliminating the classes that appear as noise.
use a much smaller box, at least for early 2D stage. ~half of this.
What is the detergent and detergent concentration?
oh good point!! this looks like DDM.
hei, I use GDN, 0.01%, does it looks like GDN micelles ?
What is the molecular weight of the particle and maximum expected diameter in Å? If the current particles do not coincide with this, then, things are looking a bit tricky. Also, how many transmembrane helices are present? The more the better in terms of aiding more accurate particle alignment. I’ve experience with a monomeric membrane protein of ~85 kDa, 14 TM helices, with 20 kDa of overall mass outside the membrane. Using a mask during 2D was helpful (depends on the extent of particle crowding), as @Ablakely mentioned,but, also:
The above may not help in this particular dataset. I have seen this before with a collaborators sample, a ~ 75 kDa membrane protein prepped in GDN (maybe 0.01 %, i forget exactly). I did one round of 2D just to confirm my suspicions, as the particles looked to be too small and having a donut like structure. The class averages look identical to what you had. We suspected that the particles were likely to be GDN micelles. It could be the same in your case. Probably best to optimise the final concentration of GDN and sample preparation (i.e, how much did you need to concentrate the sample prior to grid making etc.,)
Hope this provides some insight. Before I forget, many of these tips are from a cryoSPARC tutorial page (Tutorial: Tips for Membrane Protein Structures | CryoSPARC Guide)
Chai
Thanks for your suggestion. my protein is around 200kDa, i think it has been concentrated 10 times before applying to grids. do you have the empty GDP micelles pictutre? i was wondering how it looks like?
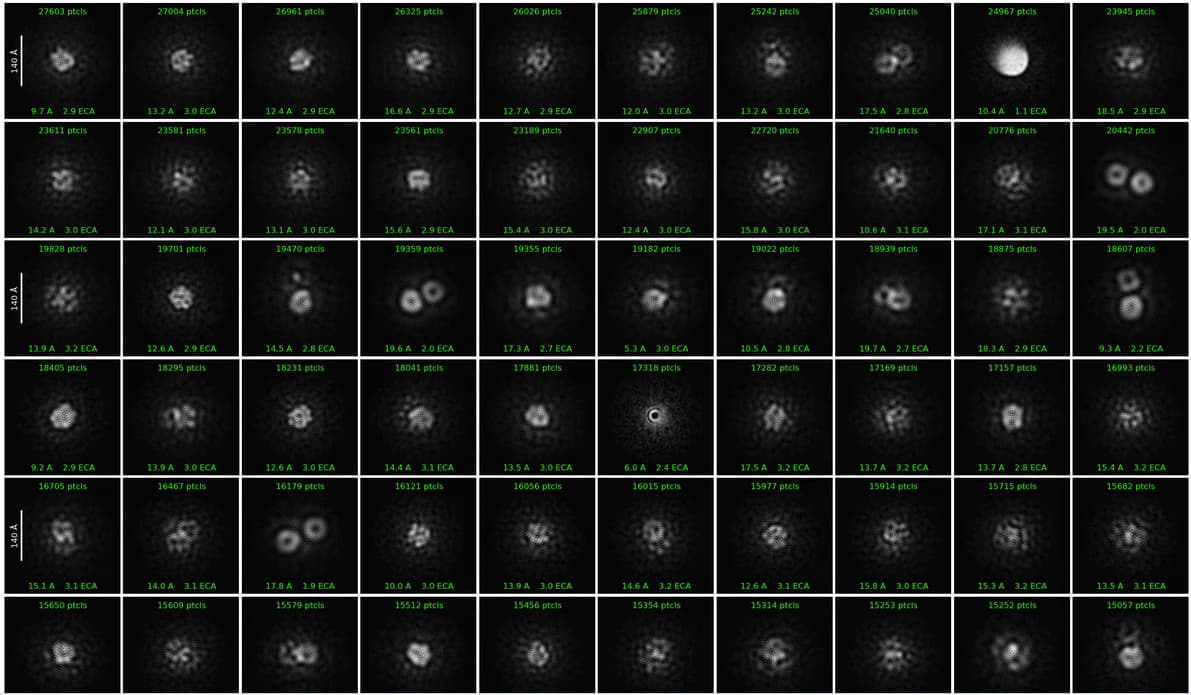
Hi,
Here are 2D class averages and one micrograph (pixel size ~ 1.2 Å/px) of the sample i suspected to be mostly micelles.


Hope it helps!
Agree this and the OP look like micelles to me