Hello, everyone! I met a problem when I do the data processing of a small soluble protein. In the 2D Classification, I can see the clear secondary structure. However, When I do the abinitio and refinement( either homo or non-uniform), the map quality was strangely bad. It looked like there is some noise “wrapping” the protein. I guessed maybe the dynamic mask was too tight? But I remember the dynamic mask was very loose. So I didn’t understand what happened. I attempted many methods, such as:
Ab-initio (12 A) - Homo Refinement - Non-Uniform Refinement
Ab-initio (12 A) - Non-Uniform Refinement
Ab-initio (6 A) - Homo Refinement - Non-Uniform Refinement
Ab-initio (6 A) - Non-Uniform Refinement
Or Ab-initio 4A…
All parameters of Homo Refinement or Non-Uniform Refinement were default.
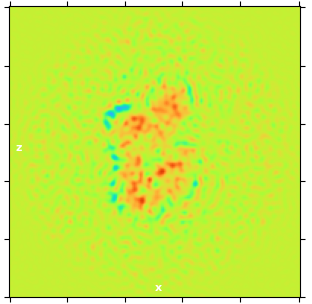
There was a 2D picture of one refinement result. You can see the noise in the picture.
These refinement jobs worked well in many membrane proteins. However, I met this problem in the case. Can someone tell me the reason and some possible ways to resolve this?
These artefacts commonly occur when the mask hugs the protein too closely and the width of the padded edge is too small for the dataset resolution. You can try manually making a mask using the Volume Tools job from the Ab-initio volume with a very soft falloff by increasing the pad width. The dilation width is less relevant; the padding is what you want to increase.
A good measure of how successful refinement is likely to be is whether you see secondary structure in your trials of Ab-initio. Other than that, the standard culprits apply to the loss of detail between 2D classification and ab-initio (discrete and/or continuous heterogeneity, poor orientation distribution, etc.).
Thanks for your patient reply! So now, I will re-do the ab-initio for max-resolution 5 A and do a mask for the ab-initio result with 6 extend and 30 soft. Can it work well? Or can you give me some suggest for specific parameters about it? Anyway, I will try it on my own firstly.