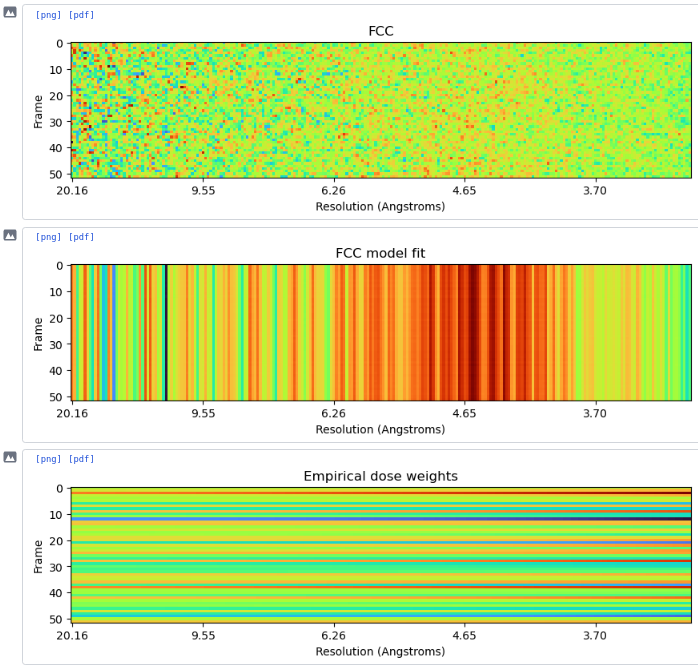
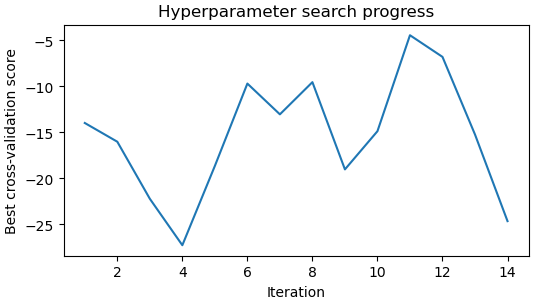
Hi all, I’m currently in the process of running reference-based motion correction on a dataset and I’m coming across some interesting plots from the hyperparameter search/empirical dose weighting. I processed my data to a 3A resolution, then ran RBMC with the default parameters. The hyperparameter search fluctuates up and down and never really stabilises, while the dose weighting maps are just incredibly noisy and clearly wrong. See below for the plots. I ran a refinement afterwards regardless, and there is a drop in resolution to 6A.
I have a feeling I know the problem, but I’m not sure how I can get around it. The data were originally processed in Relion, and the picks were then imported. Therefore, when extracting I needed to flip the micrographs in Y first so the positions were correct. I’m guessing that the RBMC doesn’t take this into account and the parameter searches are then done in the wrong place. Does this seem reasonable, or is it possible that something else is causing the problem?
Hi @mathewmclaren, thanks for your question! This RBMC result is indeed unusual.
Generally, it is not necessary to flip Y upon extraction of imported particles from RELION as this is handled behind the scenes by CryoSPARC.
I was able to reproduce similar RBMC strangeness when my workflow included an (unnecessary for my data) Y-flip during extraction of particles from a RELION star file, so this is consistent with there being an incorrect flip somewhere in your pipeline.
The way that I would typically go about the process of getting particles from a RELION star file to use in RBMC is as follows:
- Run Import Movies, Patch Motion and Patch CTF
- Import the star file, connecting the micrographs from Patch CTF and using the following:
- The star file location in the ‘Particle meta path’
- The RELION directory folder or location of the particle stacks in the ‘Particle data path’ (optional)
- Remove leading IUD input exposure file name: true
- Length of input exposure name suffix to cut (for example 31 to remove _patch_aligned_doseweighted.mrcs)
- Length of rlnMicrographName base name suffix to cut for query (for example 4 to remove .mrc)
- Run Extract mics. With ‘flip mic. in y before extract’: false
- Refine the particles in Homogeneous or NU refine (preferably with minimise over per-particle scale: true)
- Input these particles to RBMC
- Re-refine the RBMC’d particles.
If there is something about your workflow outside of CryoSPARC that necessitates an additional Y-flip, we would recommend that you use cryosparc-tools to flip the particle coordinates in Y after importing them, and before extraction so that you can go ahead with any downstream processes including RBMC without needing any further flipping. The flipping can also be performed after refinement if you have already refined particles that were (correctly for your data) extracted with a Y-flip in the micrographs.
You can find a cryosparc-tools script here that will flip particles in Y, and the usage (inside your activated cryosparc-tools environment) would be:
python flip-particle-y.py {PID} {WID} {JIDs}. If the flip is performed on freshly imported particles then an additional argument of --particle-output-name imported_particles needs to be added at the end.
I hope that solves this issue and that your next RBMC is more fruitful!
2 Likes