I’m trying to understand how the particle alignment process works during ab-initio reconstruction. Specifically, I’m looking to understand how “subunit 1” is assigned for the alignment and reconstruction and whether this could be different between each particle.
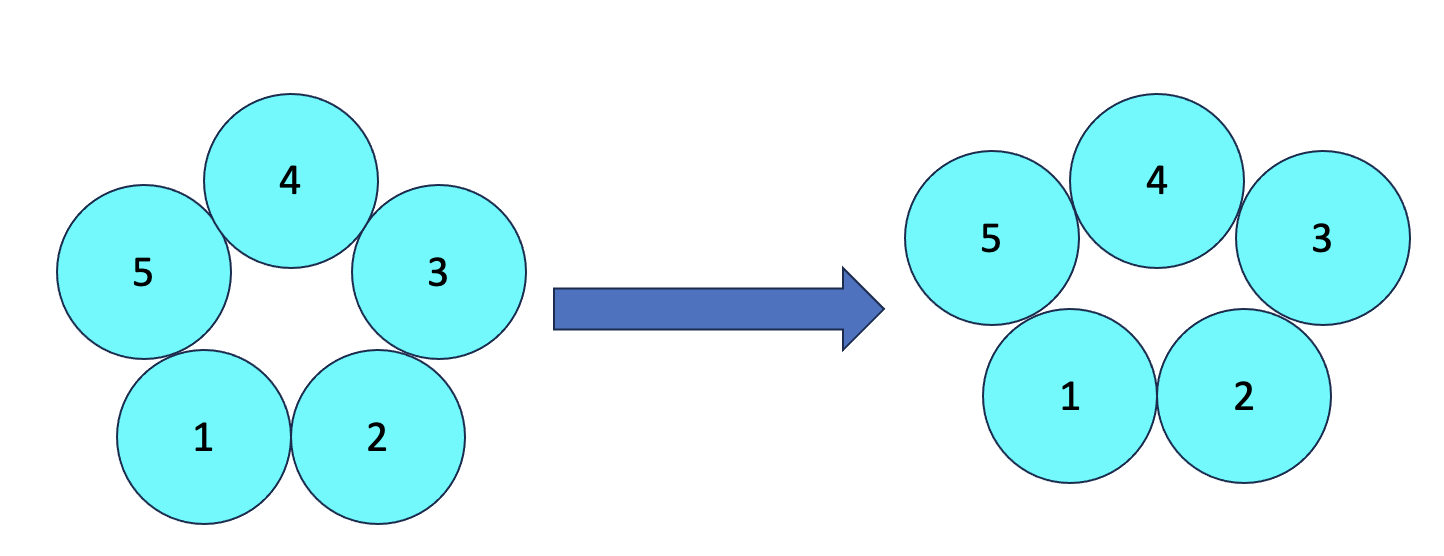
For my example, I am working on a pentameric protein that is asymmetric, with 2 subunits moving outward and 3 mostly stationary compared to the symmetric state (Think pentagon shape, with each subunit in the corner vs. trapezoidal shape in the asymmetric state or look at the image below). If subunit 1 is assigned differently between particles, the alignment would likely be off and lead to poor averaging and should lead to a loss of “features” in the “outward” subunits.
Is there a way to verify that the alignment is done correctly or a way to alter the alignment “settings” during the ab-initio reconstruction to see if the reconstruction improves?
I have a recent example that I hope will help. I was processing a dataset with two types of filaments, one with three strands and one with four. In both filaments there was a common pair of strands. When the filaments were processed together in a single reconstruction, those two strands refined well, while the other one or two strands were significantly averaged out. I could then separate the strands using one of a few different strategies to process each state separately. I would think a similar thing would happen with your data set assuming the quality of the data is good.
It’s worth pointing out that those common two filaments are not assigned in anyway, the four strand filament is symmetrical so any of the four strands could be considered subunit 1 in your example. I imagine if there are two different subsets of particles, one with subunits 1 and 2 aligned with subunits 2 and 3, for example, in your reconstruction you should be able to sort that out with 3D classification. But assuming you have two different complex states, you should be able to do an ab initio with at least two classes and separate them for independent processing. You could then use volume alignment tools or something like chimera (vop resample… after fit in map) to align the volumes to each other however you prefer.
These are good questions about particle alignment and pseudosymmetry! Let’s first talk about the basics of particle alignment in cryo-EM, and then I’ll provide a few suggestions for how you might want to proceed with your sample.
Terms
You’ll often hear people refer to symmetries using their point group. For example “C5 symmetry” describes a particle that looks the same when it’s rotated by 1/5th of a turn around the Z axis. Your particle sounds like it’s pseudo-C5 symmetric – it “should” be C5, but there’s something about it that makes it asymmetric. Asymmetric objects are equivalently called C1 symmetric.
How are images aligned
In brief, during each iteration of Ab-Initio Reconstruction, we try to find the rotation and translation that makes the current volume (the “reference”) match the particle image the best. The exact math behind it isn’t especially important right now – the important things to note are:
Ab-Initio Reconstruction uses a slightly different algorithm than other ways of generating a volume. In general, it’s best to move on from Ab-Initio once you’ve got something somewhat believable-looking. I wouldn’t worry too much about optimizing Ab-Initio and instead move on to Homogeneous or Non-Uniform Refinement.
As your map gets better your alignments improve, which then further improves the map. In the early stages of a refinement, your molecule might look exactly C5 symmetric, but as the refinement proceeds you may gain the ability to tell which positions have moved out from the center
If you’re interested in learning more about the alignment process, we discuss it in a recent workshop video you can find here. More information about Ab-Initio specifically can be found in its guide page.
How can you improve the alignment of your particle
I think in general your sample might benefit from the procedures we outline in another workshop session, which you can view here. That session covers a few different ways you can break a particle’s pseudosymmetry. TRPV5 is C4 pseudosymmetric, but the same general principles apply! You might also want to check out the Symmetry Relaxation guide page as that’s a technique that might help you out.
Please let me know if you have any more questions! In general, the more we can see what your data and your maps are currently looking like, the more we can help. Screenshots are always welcome, if you can share them!