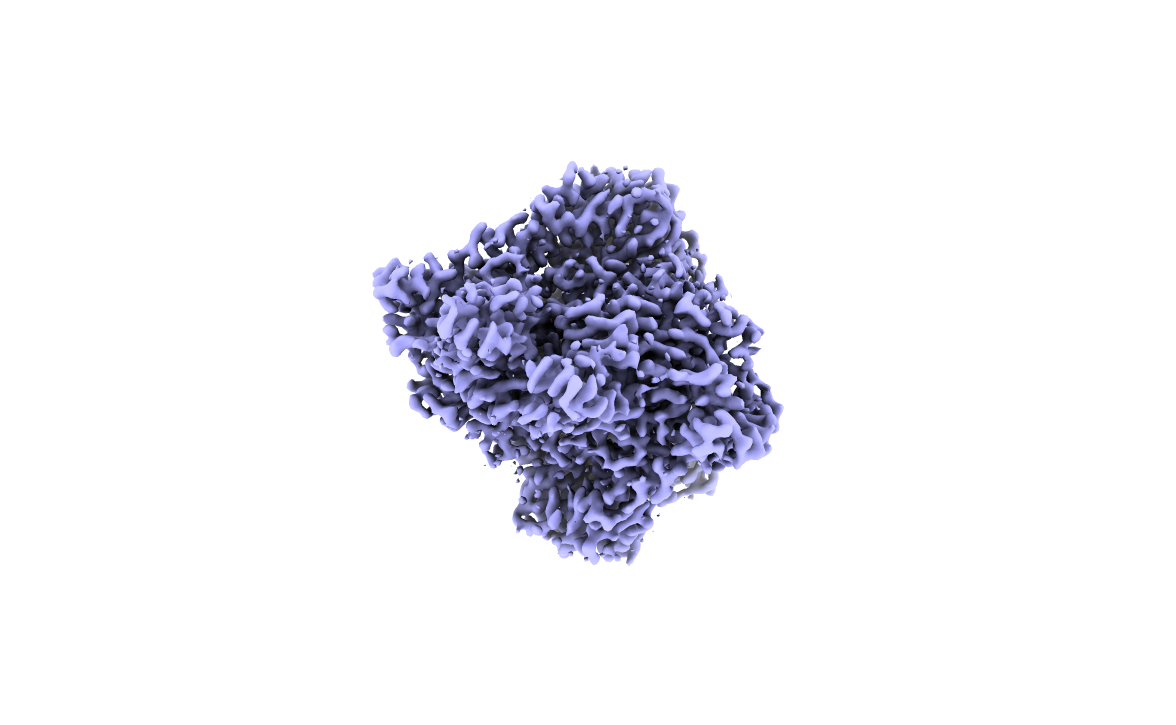
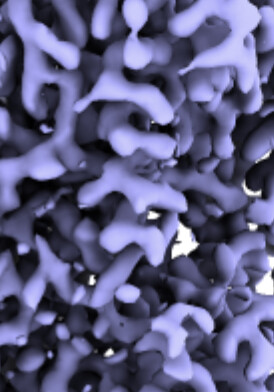
I have got 3.0 map of RNA helicase [(approx SIze is 120 kDa homodimer (58 kDA monomer)]. I have tried to fit model but I am not getting solution for fitting. I am now confused whether map which I got is correct whether it belongs to my interested molecule.
I am stuck here, any suggestions are most welcome. It will be really great. I have attached the snap of
If this is not (the whole) issue, then it is possibly a contaminant. Just glancing at the map I don’t see obvious RNA, but maybe it is more clear from another angle or in the 2D classes?
If not, try Modelangelo + Foldseek with the flipped map to see if you can identify it?
This is clearly also a similar sized dimer. Strong likelihood it’s your correct protein. Try fitting manually in low resolution maps before attempting automatic fitting to high resolution. Check alphafold2 PAE (along with good old intuition) for subdomains Vs hinge regions and imagine that domains can be rearranged.
@Mark-A-Nakasone@olibclarke
I found out the problem. This is E coli 2-oxo glutarate dehydrogenase P1 element. It is contaminant protein got selected over my protein. I used Model Angelo to trace the C-alphas from E. coli proteome.
I used 110-150 nm blob size for particle picking because my protein size is 110 kDa. but somehow this got picked up.
Currently I reduced the radius of blob to 50-80 nm. I got around 12 million particles. I have put 200 classes, batch size 400, iteration 50. but classes are hazy and of low resolution. In this 2d classification only 2 classes belongs to contaminated protein were showed up (approx. 1.5 lakh particles). But rest classes are hazy. Is this because of the particle size is small?
@Mark-A-Nakasone Yes it is A. Sorry for typo. I have attached the image for parameters used for 2d classification. Rest of the parameter I used with default values.
Try applying a smaller circular mask during 2D (e.g 100Å if the max dimension of your target is 80Å). However, I would also consider the possibility that it is just not there, in which case you will need to revisit sample preparation.
@olibclarke Thank you. I will try with mask. Yes, I will try this one because I have purified His-tag protein and after two rounds size exclusion chromatography, I could see band of the protein at respective size. But yes if I wont see any proper map, I will revisit for grid freezing.