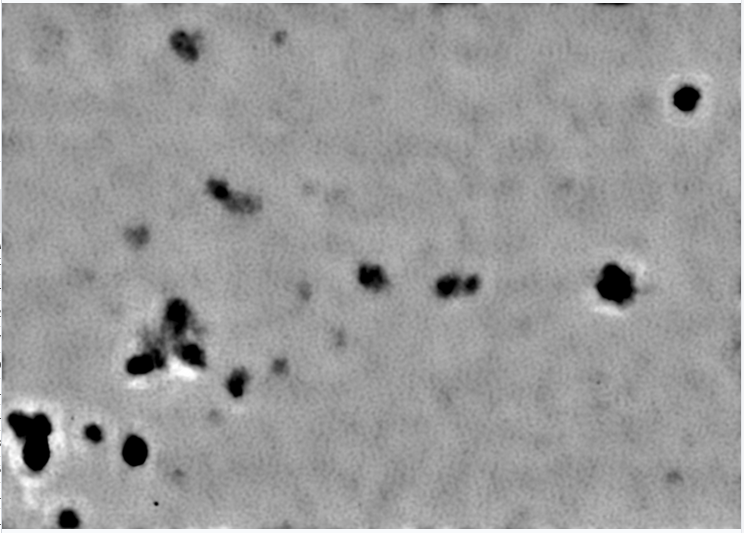
I am working with a small protein and it exists as a dimer in solution (128kDa), I collected krios data and using its homolog structure, measured the size in chimera, the maximum size is 11 nm across the diagonal. I am new to cryo-EM and i am finding it difficult to locate the protein in the micrograph. And the 2D using blobpicker gave only the noisy fuzzy 2D. Any recommendations are highly welcome. Thanks in advance.
What magnification was that micrograph taken at? That will at least give us a rough idea of how large your target might be on the micrograph…
Try denoising the micrographs (train the model, don’t use the pre-trained one! Increasing training iterations might help also) and see if you can see anything more convincing on the micrographs, because there is nothing on that micrograph which looks promising to me.
Maybe your protein stuck to the grid. Lots of things to try to resolve that, carbon film, blotting time/force… if protein doesn’t like being in the holes, I usually try carbon film first now as it usually works reasonably (but there may be other issues to contend with too - seeing your target is the first step though. )
you also need to flip your gain correction, there is clear vertical striping.
failing in iteration 11 suggests the job finally added and tried to use a particle that is bad. check that the NaN errors is part of the corrupt particles check, and perhaps try flipping the gain and then reextract.
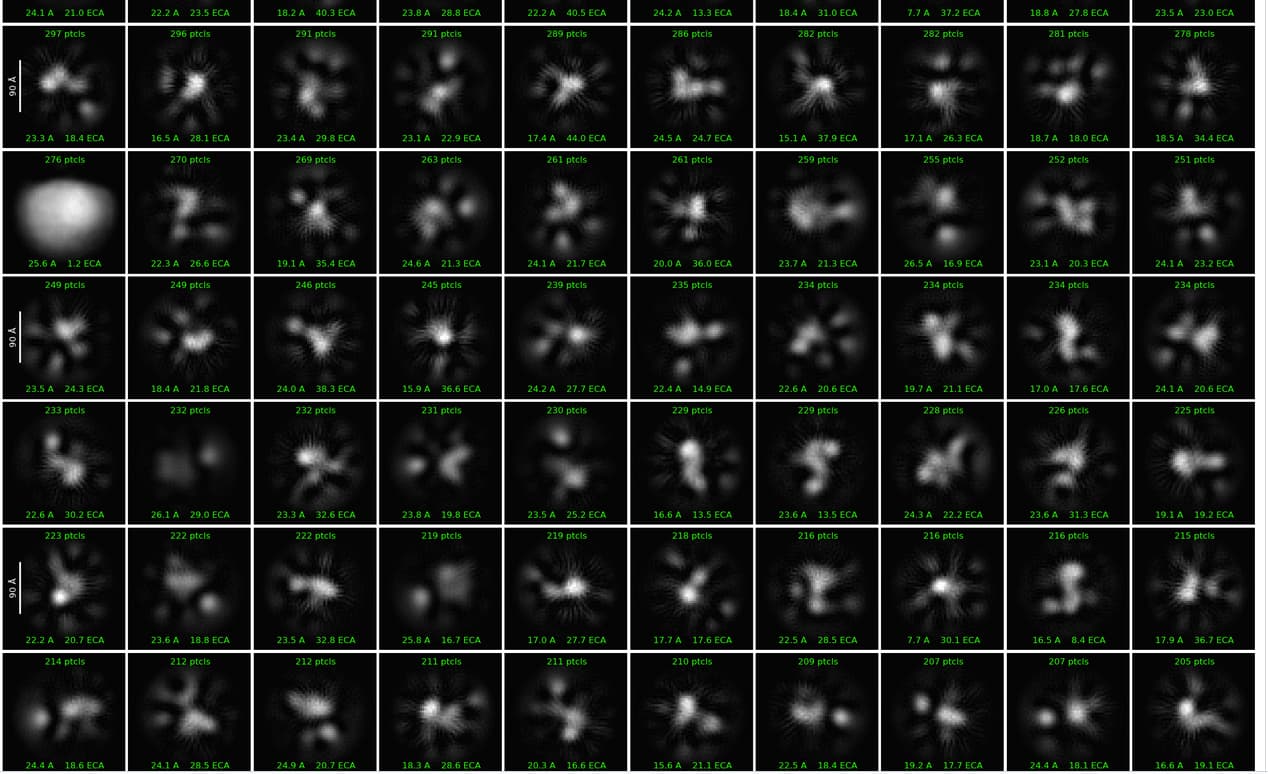
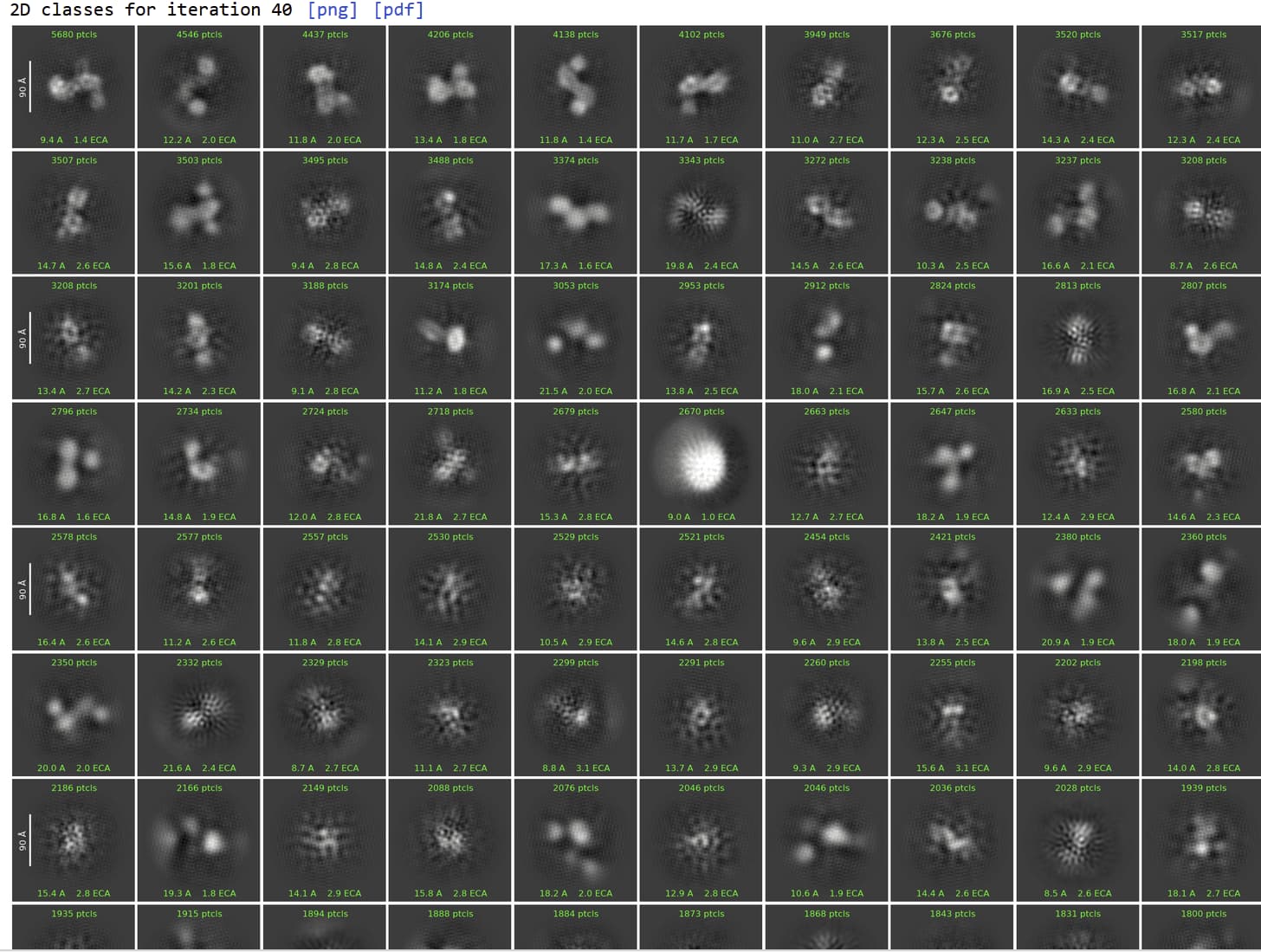
So, I tried all these things and got the 2D classes. This is a small dataset of 400 micrographs and I am doing this to get a sense of the protein. A few classes look like they are monomers of the dimer. How to clean up the 2Ds? Any suggestions?
And Thanks for the previous suggestions, helped a lot.
You don’t really have enough good particles to continue cleaning up the 2D. instead I would use the information and classification you already have to improve your outcomes for the full dataset.
To get more particles: take your first 6 classes and run 2D again with batchsize 1000 50 classes, then select the best ~2000 particles and run Topaz train/extract. Can do this multiple times simultaneously, training different models for different views.
Your box size is fine for this stage.
The white circle nearly in the middle is likely the result of picking ice spots within in the micrograph and this does not always clean up well in 3D. So you could change thresholds during picking to ignore these high-contrast picks, or you could remove this class during the 2D, or you can select this 2D, run tiny ab initio, and use that reference as a junk class in het refine.
It’s not super obvious to me that you have monomer/dimer equilibrium. The monomer classes could also be poorly aligned dimers. In full dataset I would probably keep all quality particles regardless of composition and rely on 3D (het refine or 3D classification) to separate. Should do this part iteratively - as you get a good monomer and dimer model, use that to again start het refine of all particles so they can be more confidently assigned.
Given a talk I saw yesterday at a conference, I’d try taking all but the most obvious junk and running it through ab initio (maybe 6-8 classes), then feeding that through four or five rounds of heterogeneous refinement, which over time should/might allow you to tease out some reasonable looking 3D. Maybe not at very high resolution given the particle count, but you might find that it allows you to get a better feel for whether or not the 2D contains what you’re aiming for. Then try generating templates and re-picking, aiming for a cleaner pick set, which could then be fed back into heterogeneous refinement, or used to train Topaz, as @CryoEM2 suggests.