I was trying to run ab initio reconstruction for the stressosome data which the core has icosahedral symmetry, but I keep getting an wrong initial model from the reconstruction job which looks like an cakes.
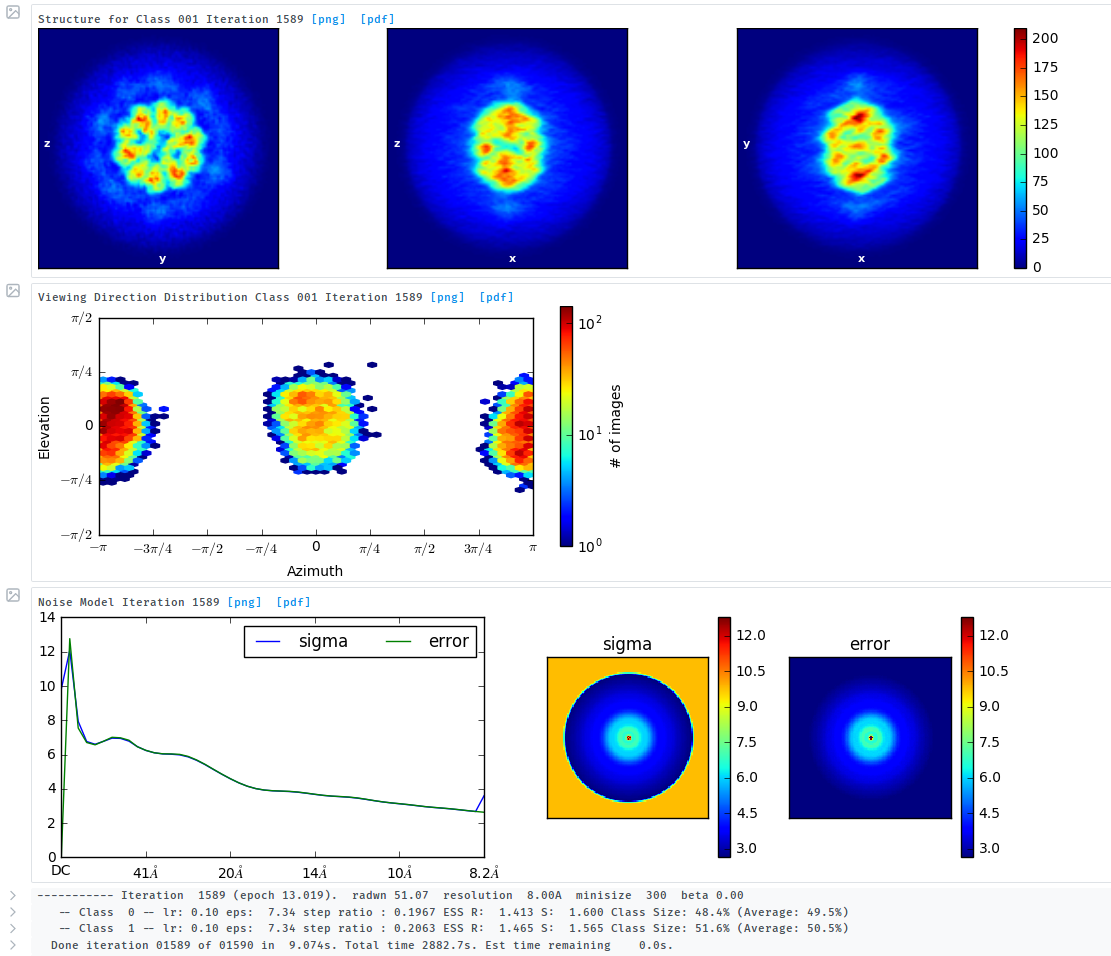
the results of 2D classification looks like this:
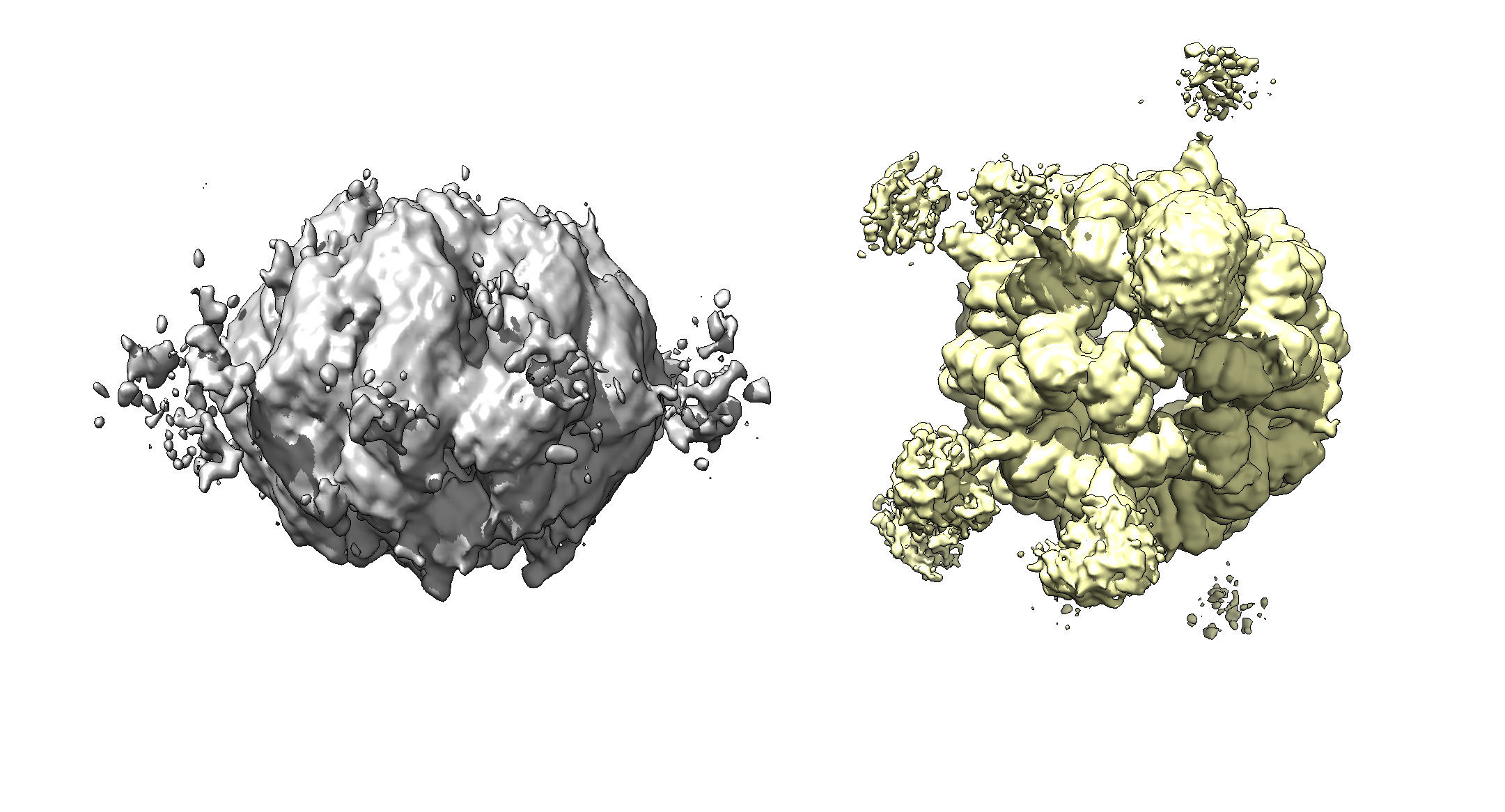
the particles are always round, this highly symmetrical core with weak singal-density turrets. but the initial model from cryosparc isnot correct and not in a ball-like shape.
Hi Lifei - I have seen the same thing with apoferritin in C1. In that case, increasing the initial and final minibatch sizes to 1000 gave a good model, while the defaults gave a “squashed” model similar to what you observe.
I recall a former labmate who was struggling on the same structure. The main problem was the mismatch between the very rigid and symmetrical core and the highly floppy n-termini (as you can see already from the 2D class averages). I would give it a try also to one available stressosome map as a starting reference, keep on from there without imposing symmetry and to treat separately the n-termini from the core.
Hi, Oliver,
Thanks for your suggestion. i tried to increase the intial and final minibatch sizes t0 1000 or more.
The generated intial models are still same-- a “squashed” core.
In the past, In Relion 3, the Ab initio reconstruction for this data always gave me the correct initial models.
as for as i know, the Relion3.0, the implementation of de novo reconstruction is similar to the one in cryosparc.
so, i am wondering that the reason generating wrong initial models in cryosparc.
because there is no particles in the data is similar like the projection of the a “squashed” core in that angles.
Hi, Edoardo,
thanks for your advice. I used Relion 3 to creat some intial models which look reasonable, then used it as reference.
there was misalignment for this highly symmetrical core with weak singal-density turrets. It is hard for the algorithm to assign the correct orientations compared the images with the map projections.
we did the signal subtration in the relion to treat the turrets and core separately then ran the local refinment for the core structure to imrpove the resolution.
Thanks for reporting. For these highly symmetric particles (at least the core in your case) the squashed result can occur due to the fact that all projections look quite similar at low resolutions, so that the beginning, ab-initio reconstruction will rather assign all particles to the same viewing direction, when the initial model is very far from correct.
Along with the minibatch size, increasing the
Initial resolution (A): to 15 or 20 A and
Final resolution (A): to 8 or 6 A
will generaly help a lot when the only alignable signal is at higher resolutions. The job will run slower but give a better result. This also works for C1 reconstructions of apofettitin.
I encountered similar problem for my particles (Oh global symmetry). But neither increasing batch size to 1000 or 2000, nor resolution limits gave me correct initial model. My question is what would be now more sensible to change further: batch size or resolution? I need to admit that models generated now are better, but still far away from being correct.
Hi,
For me increasing the
Initial resolution (A): to 12A and
Final resolution (A): to 5-6 A.
batch size: 1000,
finally, I think, it is more sensitive to the final resolution.
the initial models look very reasonable.
Best
Lifei
I am seeing exactly the same behaviour as Lifei with an iocsahedral virus. The 2D classes look nice, but the initial model is squashed. I currently have initial and final minibatch sizes set to 1000 and I am going from 12 to 6 Ang resolution. Unfortunately, these settings don’t seem to help. As I’m concerned that alignment can be skewed by neighbouring features such as carbon edges and other particles, I have been using a fairly tight mask in 2D classification. Is this same mask applied in the ab-intio job? I couldn’t see anywhere to set this in the job builder for this task, even in expert mode.