I have collected a dataset of a small membrane protein. The protein particles appear to be densely packed, but relatively dispersed.
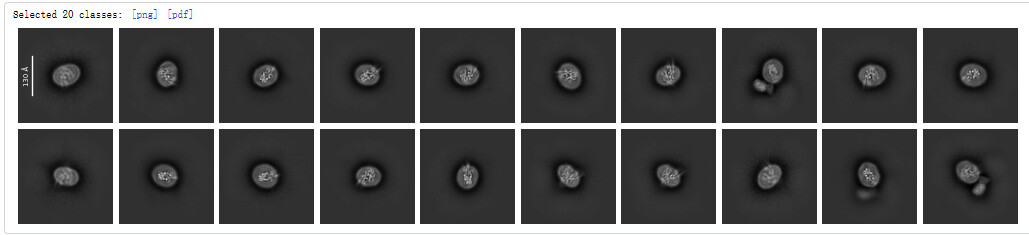
After picking the particles, I performed 2D classification.After removing obvious junk and classes with multiple particles, I performed several more rounds of 2D classification. Finally, I selected these classes.However, some of these classes belong to the top view, other belonging to the side view (such as those circled in red), in which, the transmembrane regions are clearly visible, but strangely, they have lost the extracellular alignment fiducial marker.
Here is parameters for 2D classification:
Classes : 200 ( or 300 , my total particles are among 2~3 million.)
Minimum separation distance (A) : 50
Number of online-EM iterations : 40 ( or 30 )
Batchsize per class : 1000
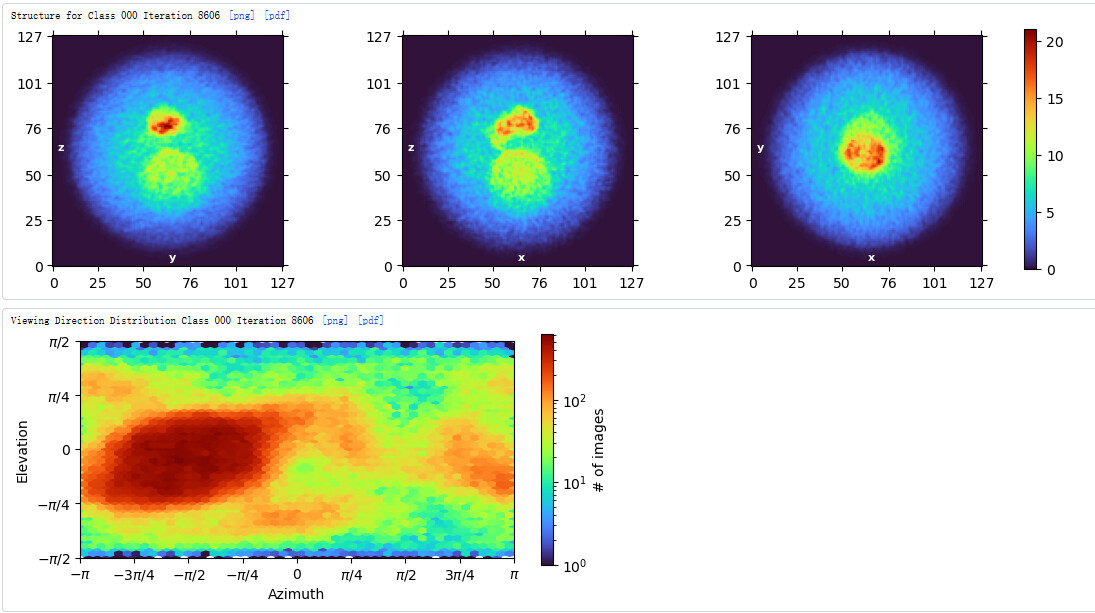
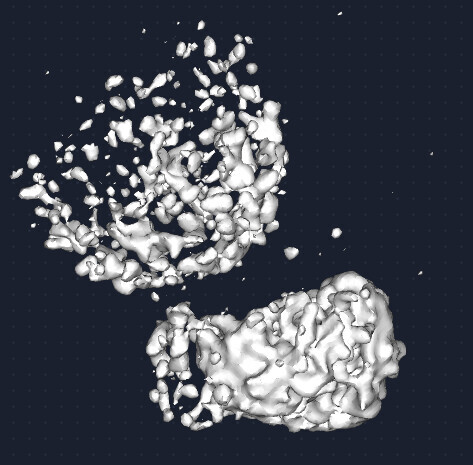
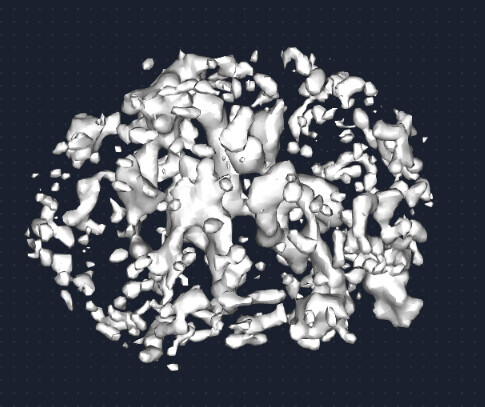
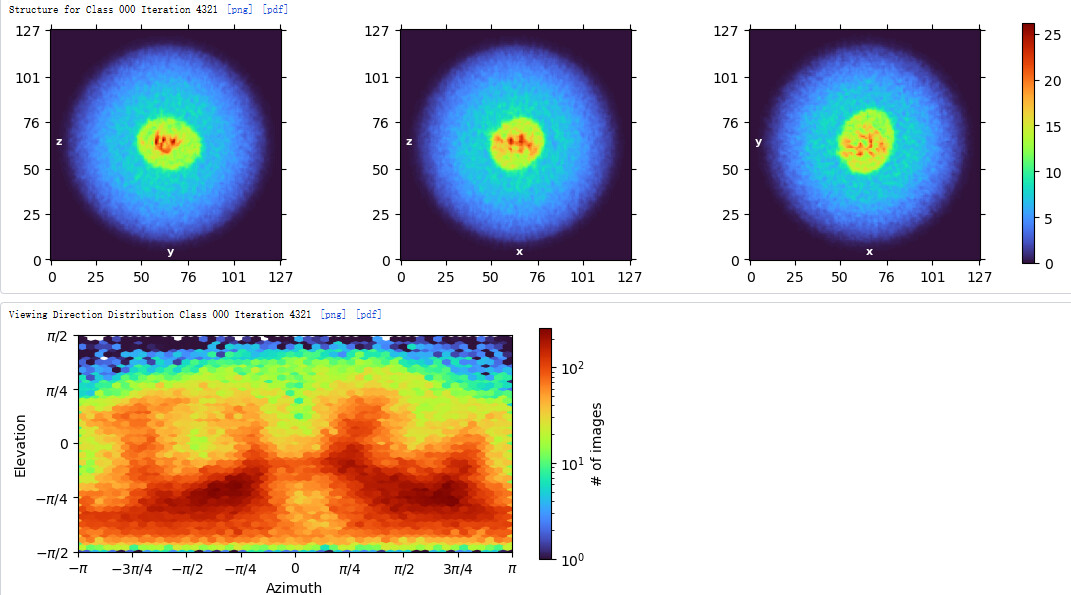
Subsequently, I used the above classes to perform Ab-initio reconstruction. But I was unable to obtain a complete map of the particle.The micelle is incomplete, and clear transmembrane regions are not visible. Additionally, from the viewing distribution plot, it seems there are dominant orientations.
Some of the classes also lack extracellular features. In these cases, it’s possible to see the micelle and a portion of the transmembrane region, but they are not complete.
Here is parameters for Ab-initio:
Number of Ab-Initio classes : 8
Maximum resolution (Angstroms) : 5
Initial resolution (Angstroms) : 16
So, my two questions are:
Why can’t the fiducial marker outside the micelle be seen in the side view of the 2D class?
My 3D results seem to have a preferred orientation. Do you have any good suggestions to solve this problem?
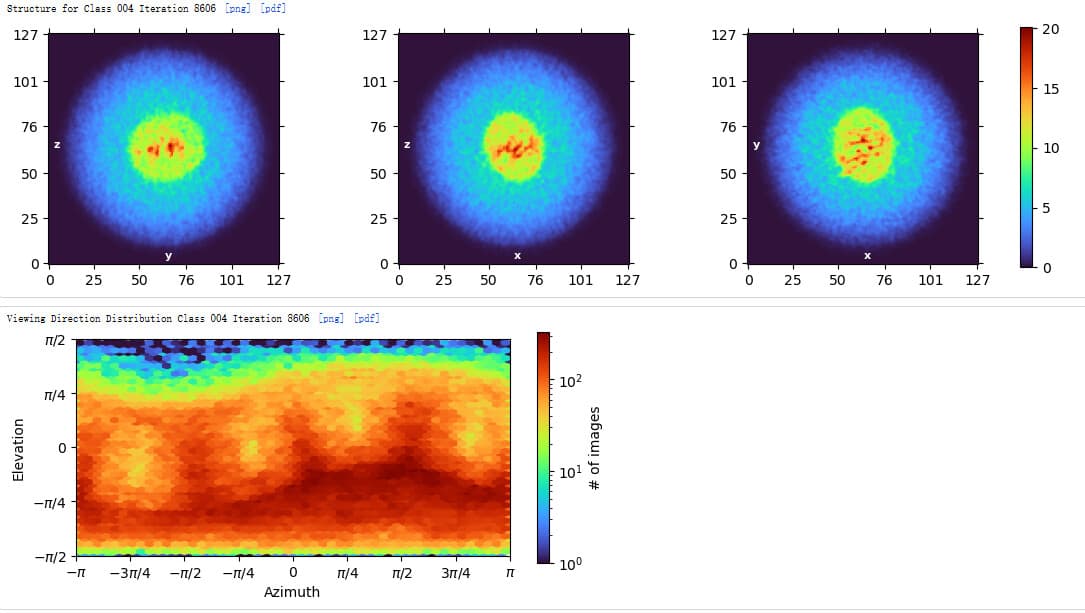
These all look like top views to me (with some slightly oblique views), no obvious side views in the classes or micrograph. I would guess preferential adherence to the air water interface, combined with partial dissociation of your fiducial.
I completely agree with your point that the 2D images all seem to be top views.
However, I would like to ask you two questions:
Could the dense particle distribution (higher protein concentration) affect the structure determination?
The 2D images do not look particularly perfect, with many features appearing quite blurry. Could this be due to the nature of the protein itself, or is it a computational issue?
Thank you for your insights!
Could the dense particle distribution (higher protein concentration) affect the structure determination?
If particles are so close as to be overlapping, or interacting in a manner that leads to partially ordered aggregates that could be an issue, but doesn’t seem to be the case here IMO.
The 2D images do not look particularly perfect, with many features appearing quite blurry. Could this be due to the nature of the protein itself, or is it a computational issue?
To me, the images without fiducial actually look pretty good, with clear secondary structural features within the TM region. The ones where the fiducial is visible seem prone to aligning on the fiducial, which blurs out the TM region, presumably because the fiducial is binding to a flexible or mobile epitope. I would try sample prep without the fiducial, and screen surface-active additives to identify something that mitigates the preferred orientation issue.
Thank you for your response!
However, I have some questions. Aren’t the classes circled in red in picture2 side views, even if they have lost the fiducial marker? I have tried using only these “side view” classes and top view classes that lost the fiducial marker for Ab-initio reconstruction, but the result still seems to have a preferred orientation.Besides, I can only see a part of TM.
No, they do not look like side views - they look like top views. See how the micelle or nanodisc is oval shaped - this is the projection one expects from the top, or near the top.
In side view, you should see something linear, that looks rather more like a membrane, with vertical banding from TM helices - that is not evident here.
Hallo Shimmer,
As Olibclarke suggested,you can try to make use of the NanoDSF to screen short chain detergent (OG,NG, CHAPSO…) to make sure the used detergent does not destabilise or disassemble your protein before making new grids.
Or, you can tilt the stage if the ice thickness is good enough and the sample prep is time consuming.
Good luck,
Ge
I do plan to address the preferred orientation issue by adding an appropriate detergent.
However,I’m not very familiar with your second suggestion. Could you please tell me what kind of ice thickness is suitable for data collection with a tilted sample stage? As Olibclarke mentioned, if the micrograph is almost entirely top views with no obvious side views, is this also suitable for the tilted sample stage method? If so, what are the principles for choosing the tilt angle?
Caveat is that there is more beam induced motion and effectively thicker ice at higher tilts, and it won’t fix the fundamental problem of your particle adhering to the air water interface, but it will recover some missing views
Beautiful top view! It’s definitely worth to try additional detergent as @geyang928 suggested. In one of my case OG improved particle behavior but CHAPSO made it even worse. So you may need to try different detergent (and concentration), collect a small data set, run 2d classification to see if there is any improvement.
Excellent suggestion. In one of my examples with a soluble protein, I encountered severe preferred orientation under native conditions, with almost all particles in side view, preventing me from obtaining an initial model. I then tried using CHAPS at a final concentration of 0.025%, which yielded top view conformations. However, when I tried β-OG at 0.02% and 0.04%, the results were not satisfactory. Could you share the concentration of OG that has worked successfully in your cases?