Hi all, I am a new Cryo-EM user and working with my first dataset.
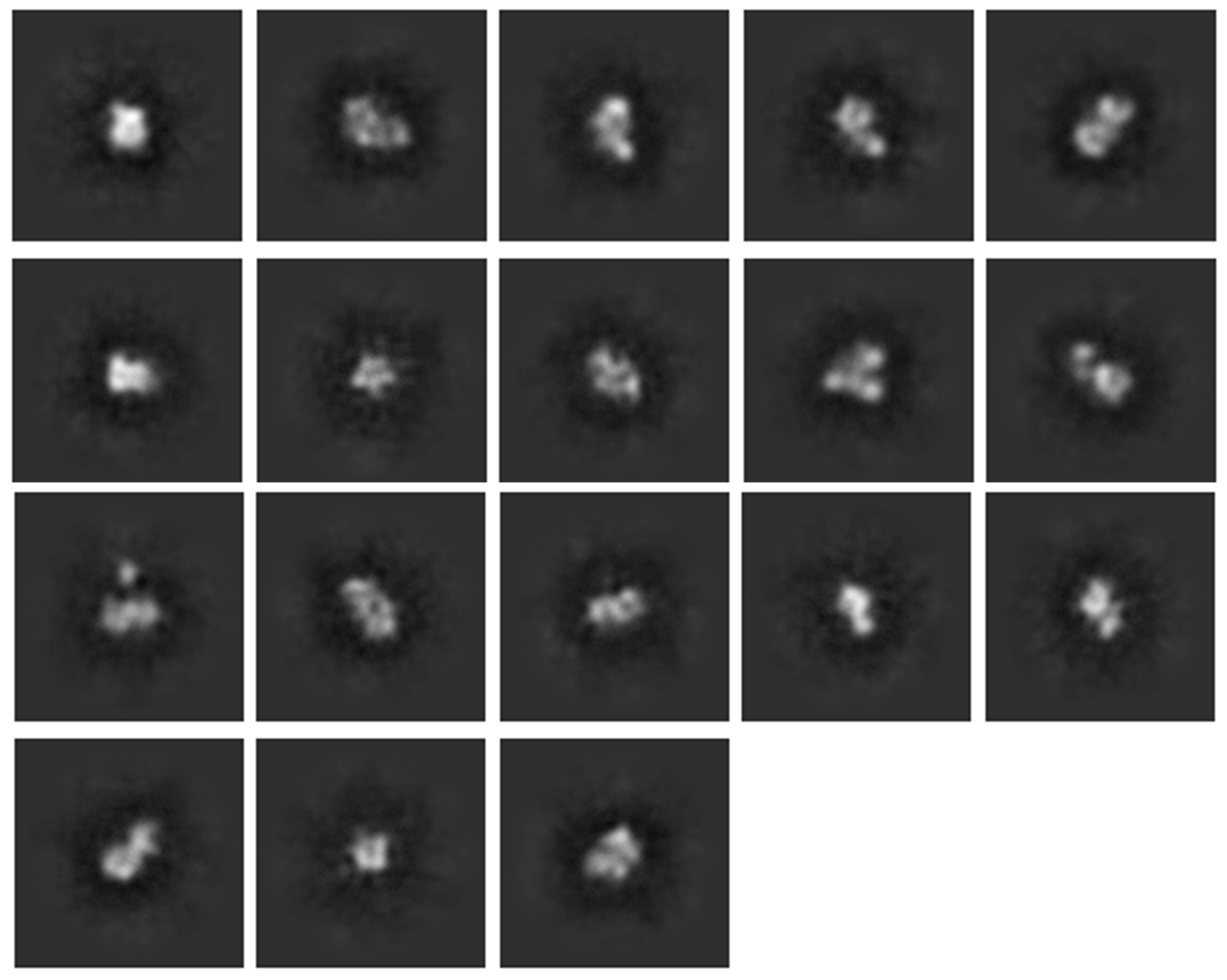
I am working with a protein which is approximately 130kDa in size and forms a dimer in presence of a protein ligand. This protein has multiple domains with long flexible linkers (longest linker 45 a.a). All together it should be 2:2 complex. The ligand is approximately 30kDa in size with its tag. The sample was purified by affinity purification followed by SEC-GraFiX-SEC. Peak fractions were pooled together and concentrated used for making UltraAuFoil grids R 1.2/1.3 using Vitrobot. I collected 4000 movie frames on Glacios microscope (200kV) with K3 camera in super resolution mode. I used approximately 3000 micrographs after curation for further processing and my initial 2D classes were very poor in resolution and most of them were off centered. Despite various attempts to center them I got only 18 classes with some definite shapes (Figure 1).
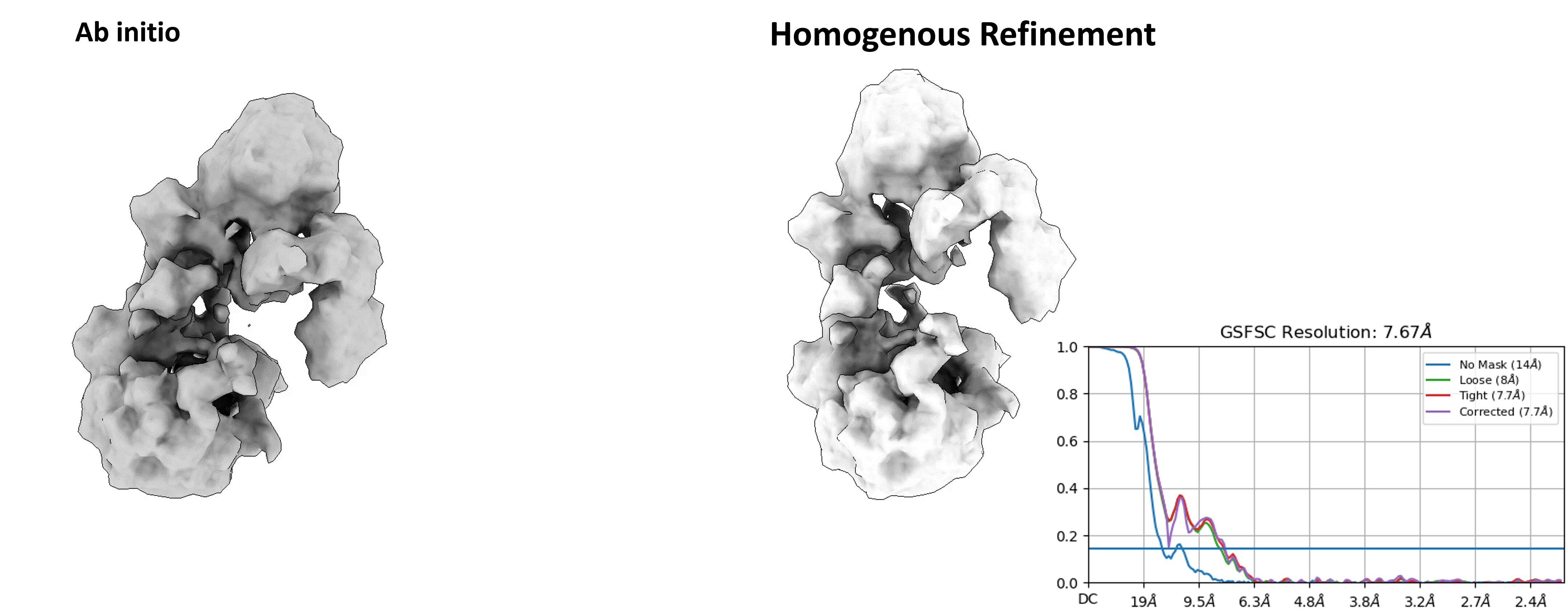
Upon several rounds of template picking jobs and using various parameters, I was able to clean it up but with a very tight mask (Figure 2). I ended up with approximately 80,000 particles. I tried generating 1 ab initio model with the 2D classes (Figure 2) and went ahead with homogenous refinement. Though the GFSC curve graph looks good (Figure 3), but the map is very broken and looks streaky.
My question are-
why I am not able to get 2D classes showing higher resolution features?
Is it possible that these 2D classes are just some misaligned particles and I have no good particles to solve the data?
Possibly, my sample is extremely heterogenous. So, does it make sense to collect more data to increase the number of good particles to generate good 2D classes and 3D models?
I have tried a lot of cleaning strategies for 2D classes previously suggested in forum like performing different ab initio classification, selecting those particles, performing 2D classification again, or performing heterogenous classification, selecting particles, doing 2D classification or trying different extraction box sizes etc. Whatever I do I always end up with very little particles in my 2D classes and Ab initio model does not looks good.
I will highly appreciate any suggestions on how to improve my data.
Thanks,
Ashish
For figure 2, it was approximately 237Å (432 pixels) and was without any Fourier cropping. The particle diameter is approx. 120Å.
Yes, I tried fitting the Alphafold model of the structure in the homogenous refined map and it seems to be kind of fitting. Which is surprising to me because after fitting it looks like a monomer to me. On SEC it was a dimer peak.
2D classification parameters for Figure 1-
All default values with number of classes - 100
2D classification parameters for Figure 2-
Number of classes -100
Initial classification uncertainty factor - 3
The mask diameter used was- 150 Å
Number of final full iterations- 2
Number of online-EM iterations- 60
Batchsize per class- 500
On the right track. Unfortunately, (and don’t tell Holger I said so), Grafix is likely a big issue. Skip it, or do the gradient no fixative. I’ve been back and forth on Glutaraldehyde, but hardly seems necessary for tiny proteins/complexes. And very often ruins high res alignment in my experience.
Topaz pick.
Apply bias (oh my!) by aligning to 10A molmap (will have to change 3D low pass filter to less) in either het or NU-refine.
Then can always clean up and sort good particles and take that particle set back to the beginning for gold-standards.
That homo refine is just model bias, it’s a parrot of the an initio which may be close or may be way off
May I ask you how to apply bias for the grafix data? My sample is also grafix and half of them I can get high resolution, but the other part some classes are rigid, while some of them seems not stable. I tried many ways but I just can’t get difference classes. May I ask you for some suggestions?
you mean for one multiprotein complex, you run Grafix procedure, and generally can solve high resolution structure for part of the complex, but other parts of the complex you cannot see at high resolution? And you cannot seem to separate the compositional and conforamtional heteroegeneity?
if that’s the case, then you do have all particles well-aligned to a single reference via homogeneous refinement or non-uniform refinement enabling high resolution. Hopefully you have a LOT of particles. I would take those particles/reference into 3D variability filter resolution 4Å, then 3DVA display “simple” mode filter resolution 4Å using component 0 for one job, component 1 for one job, component 2 for one job. this will let you see/morph maps the different kinds of motion/heterogeneity and make decisions about how to proceed. Would for instance then run 3DVA display again with the component(s) that seem most important but this time cluster to 20 classes.
Easier; 3D classification with 20 classes, resolution 14, initial o-EM 0.8 instead of 0.4, class similarity 0, turn off density check for convergence toggle, turn on output results at each full o-EM.
By bias, I simply assumed the author had crystal/alphafold models for the complex, could go to chimera and use molmap command to generate densities that correspond to the full complex or subcomplex models, then import those and use them as references in het refine or NU-refine with initial lowpass filter changed to 10 or so. I was assuming their refinements were failing not for lack of good particles but for lack of good starting model for alignment.