I’m working on a small (~140 kDa) flexible protein for which no experimental structural data is available. A sample of my 2D classes is below. I’ve tried multi-class ab initio refinement and it always splits the particles into approximately equal classes (5-10% variation in population).
When I try heterogenous refinement the resulting classes are still equal in population. Homogenous or non-uniform refinement on any of these volumes leads to models which are ~6 A in resolution but don’t have any secondary structure features. I also tried a single-class ab-initio refinement with similar results.
I’ve used the preset parameters for the refinements, and for the ab initio reconstructions I changed the initial and final resolutions to 7 and 12 A respectively, and initial and final minibatch sizes to 400 and 1200 respectively.
My current idea is to continue sorting through the particles using 2D classification (at this point its getting hard for me to distinguish good from bad particles though). If anyone else has any insights or suggestions, they are deeply appreciated!

Oh boy… it seems very hard to align those… would you mind posting one image example?
Hi, how many particles do you have to work with? A lot of the classes in your screenshot do not even have 1000 particles. If you’re working in the 50-100k range (prior to 3d classification) with a particle of that size (and that apparent flexibility/heterogeneity), I can imagine that you will just run out of particles/signal during your classification attempts.
Hi Carlos,
Yes it doesn’t seem to have any good features to align on unfortunately! The image below is of one of the ab-initio reconstructions.
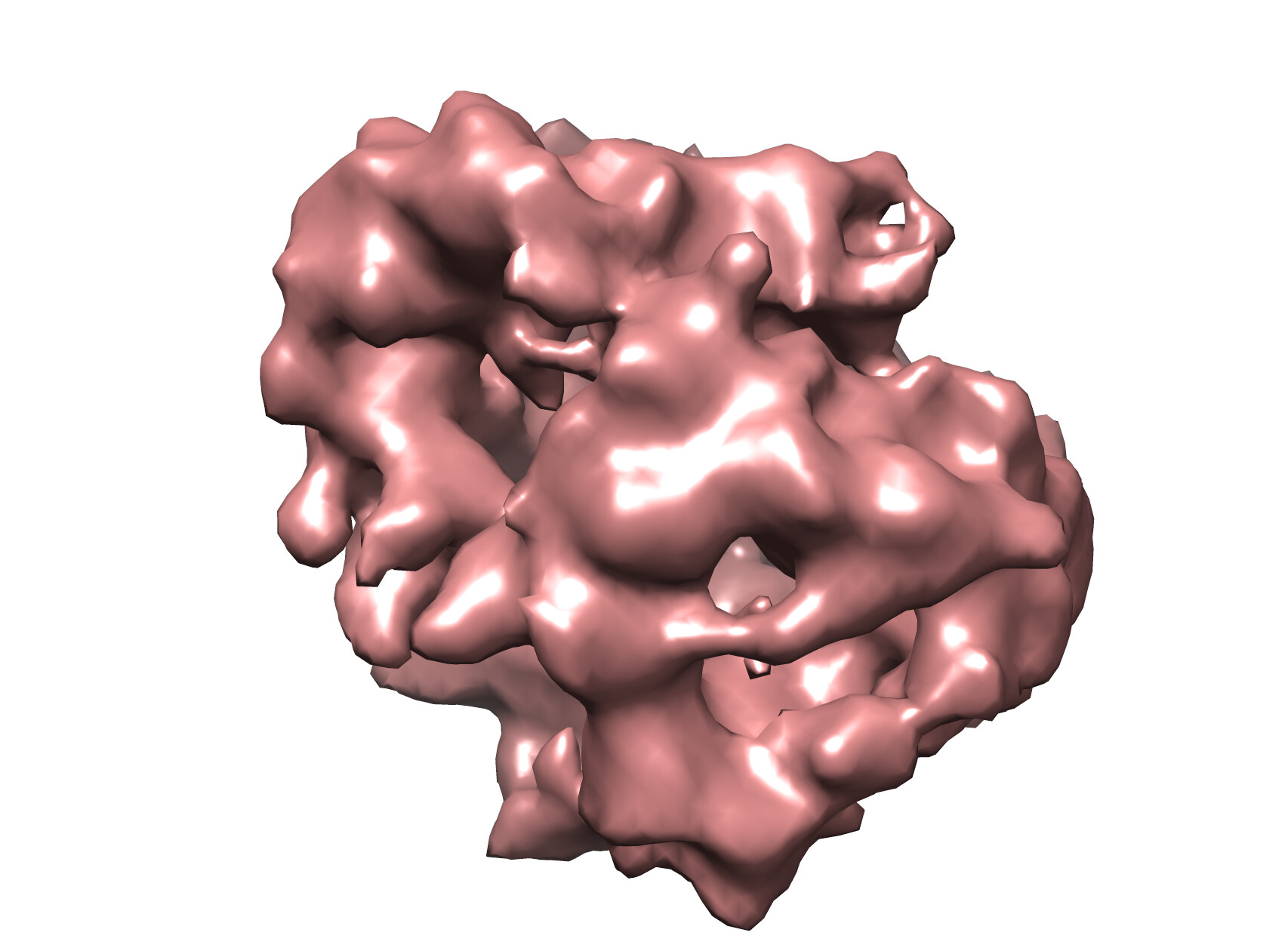
Hi Moritz,
The example I’ve posted has about 100k particles, but I’ve gotten similar results with ~200k particles as well. It’s quite possible I need more data/particles though, thanks!
These 2D classes look a bit odd. What settings are you using? If you are using clamp solvent, I would turn it off, and try with Force/max off and an increased number of iterations (40-60) and larger batch size per class (maybe 400)
your box size may also be a bit large for an initial round of classification - what box size are you using?
I used ‘enforce non-negativity’ for the classes I showed above. I’ve attached an example without that option below. I’ve consistently used Force/max off since it looks overfitted/streaky with it on. Using more iterations seems to also lead to overfitting, but I could try it with larger batch sizes, thanks! I’m using a box size of 240 currently - I was worried that I might cut off some extended conformations if I went too small. Thank you for your suggestions!

I wouldn’t use enforce non-negativity - leads to weird artefacts. I would double check your picking and data collection parameters - is your picking good, is the ice too thick, what do CTF fits look like?
For this dataset I filtered CTF fits in a range of 2.1-3.1. My defocus range, relative ice thickness etc were all within standard parameters, though it’s possible the ice is still too thick. I picked using Topaz, and I trained the models using a set of blob-picked and sorted particles. I have tried just blob-picking as well, in terms of alignment of the 2D classes Topaz does seem to work better. I’ll keep the artefacts issue with non-negativity in mind, thanks!
can you show a picked micrograph with picks illustrated using inspect picks and box size 20? this is extremely interesting, are you sure the protein is 140kDa via gel/SEC? should see helices and domains in 2D. what does alpha fold predict? you could use that as template pick or 3D model if you are from an experienced lab and know how to handle bias. otherwise it might be a floppy mess and you need some structural rigidity, maybe glut crosslink? 2.1Å CTF is amazing in any of the programs, there must be 1000 picks per image? or on carbon? it’s possible but not probable that 10x the data will latch on to something in 2D, no guarantee it will be able to solve 3D problem of finding a core for high-res alignment
Hi, yes I’m sure the protein is ~140kDa, using both gel and SEC. I’m hesitant to use Alphafold prediction as a model since this protein doesn’t have any close homologues, and the tertiary fold of the predicted structure looks improbable (domains packed in a globular fashion with long unstructured linkers encircling it). I am considering cross-linking in the next iteration, thank you for the suggestion! Below is a micrograph picked by topaz. It does pick ~900 particles per micrograph.

if you measure the size of your particles on the micrograph, how big do they seem to be? My impression is that they look smaller than 140kDa - I would consider the possibility that your protein has fallen apart during vitrification, and think about either the use of surface-active additives or mild crosslinking.
That’s definitely a possibility too, albeit a very depressing one. Cross-linking seems to be one of the consensus ideas here, thank you!
I think alpha fold is correct, there are several structured domains and their association with each other is either globular or random, so in ice and with AWI compounding the problem you likely see 50kDa tethered domains and are trying to solve several independent tiny structures. Does anyone else in lab have a ~150kDa protein for comparative analysis, along with Oli’s suggestion to measure. b-OG, LMNG, Amphipol A-85 are options. did you target a single square of relatively thick ice? same particle image in micrographs for that one?
Yes I would assume it’s more like beads on a string and so I’m looking at individual domain densities with invisible linkers. I don’t have anything on hand for a comparative analysis, but I can ask around. I did screen some grids with relatively thick ice but I couldn’t see any particles in those at all.