I’m working on a trimeric protein that has been purified in SMA liponanoparticles, but unlike the usual transmembrane proteins, mine is anchored to the membrane via a re-entrant helix that enters and exits on the same leaflet. I have a dataset that I was able to pick ~500k particles from, and curate them down to ~100k via 2D classification.
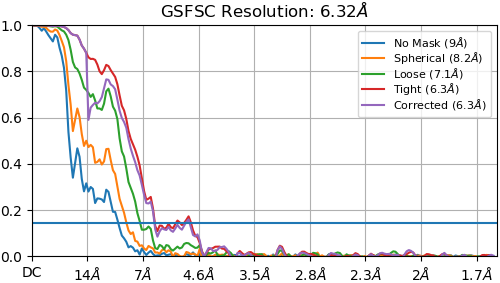
If I include all 100K particles, the best 3D reconstruction resolution I can get is ~15 Å. However, if I pick only the best looking class from 2D classification, I can get a reconstruction that is good to ~7 Å with symmetry enabled using non-uniform refinement (see below). However, this only contains ~2300 particles out of my original 100k.
I’m inclined to think that I can boost my resolution by including more particles, but I’m not sure why that’s making my resolution worse in this case. Does anyone have any tips for a situation like this?
Native nanodiscs can be extremely heterogeneous, so that may be the fundamental issue. Most likely you still need to optimize the biochemistry to get to high resolution. I see that in your 2D classes, there is one very good class which also contains about 2,300 particles. How dense are the micrographs? Is there a lot of junk everywhere?
Try the standard 2D advice - 40+ online iterations, 400+ batch size, limit alignment resolution to 12A, 100 classes. Use your best reconstruction to seed heterogeneous refinement of several classes against an earlier “non-junk” 2D selection (perhaps using downscaled particles). Look at your maps and orientation distribution and think about if this is a major limiting factor as well. Ultimately, 500k initial particles is not really that many and you may need to increase the data size ~5x on top of the above parameters and any biochemical improvements you can come up with.
Edit
PS adding more particles is making your resolution worse because they are not good/averageable particles except for those few you are pulling out.
These are affinity purified and look quite pure by SDS-PAGE and mass photometry, but there’s a chance there are some empty smalps that came along. Conformational variability is also a definite possibility.
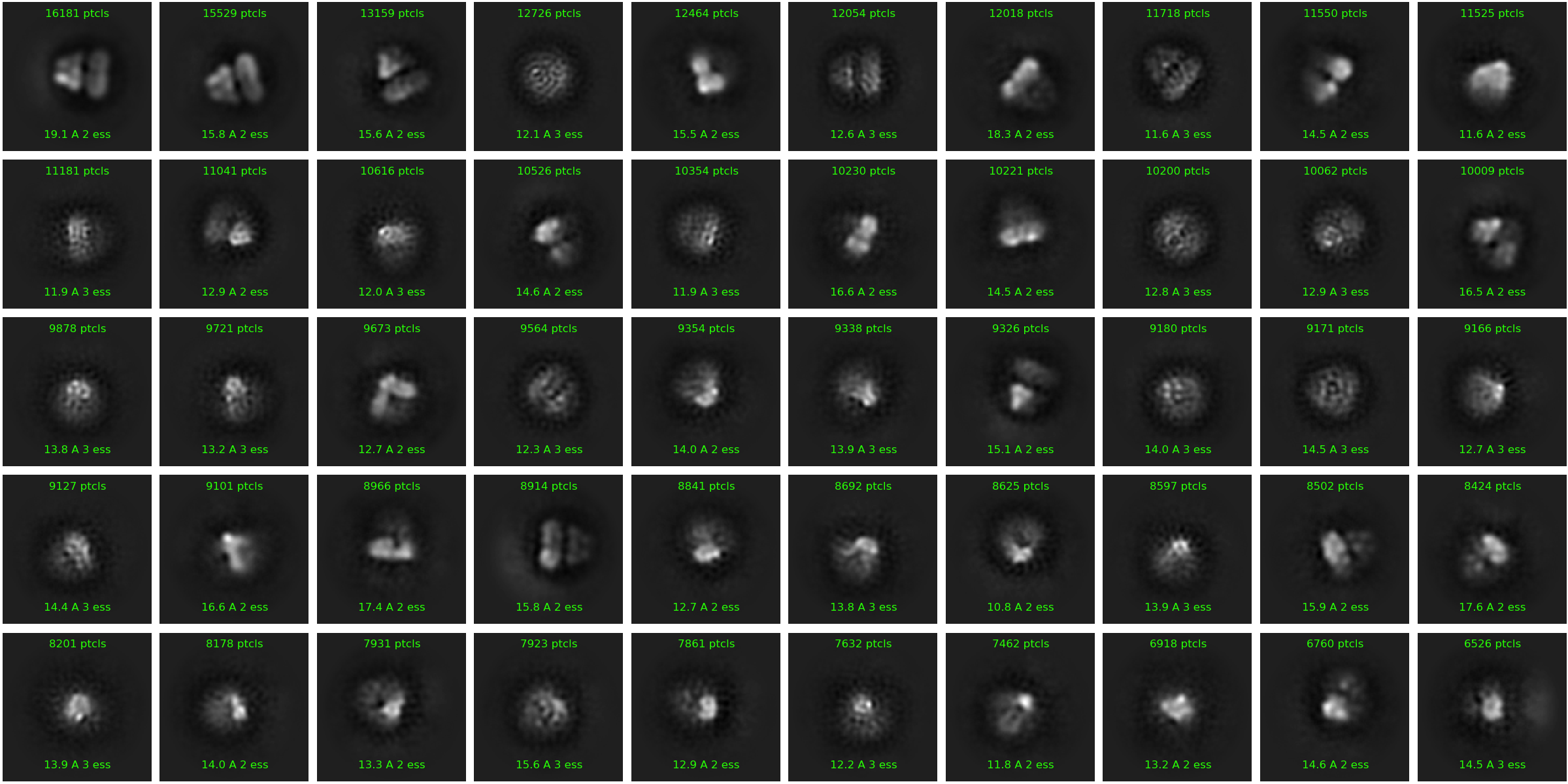
One of the things that I’ve noticed in 2D is that for many of the classes the particles seem to get stuck centered on an “edge” of either the lipid or protein portion of the particles (row 1 image 1, row 1 image 8 in 100K particle image). I imagine this is tough since the center of mass for these particles is roughly between the disc and soluble domain, and has little density in the side views I’m capturing. Right now I’m using a ~350 Å box size, I wonder if increasing that would help.
Hi,
in your 100K 2D classes, you have beautiful one in the middle where secondary structures are clearly apparent (same amount of particles as your 3D reconstruction), while all the other classes are more blurry. My hunch is that your SMALP is moving around your protein, and even if the views look similar, they are different enough to preclude good alignment.
Maybe you can continue on with the refinement with the 100K particles, and using a mask including only your protein. You will dilute the SMALP signal but I think that’s not what you are interested in.
Best of luck
Vincent
Yeah! I tried to explain in the OP, but the 3D reconstruction I posted is generated from only that excellent 2D class. I suppose the symmetry is helping me out a lot given the small size of the class. I’ll give masked refinement with the larger particle set a go.
Forgive me for stressing this so much, but don’t fall into the trap of thinking the biochemistry is good enough. The biochemistry is never good enough and is always the primary limiting factor unless the microscope was poorly aligned or there are severe orientation preferences. I have learned this the hard way multiple times now, and seen other people waste a lot of time thinking better microscope or software use would help them. Have you tried SEC? If you lose too much material, you can try adding more charge screening against the highly negative SMALP. Typically SEC dramatically improves monodispersity in cryo-EM. (An alternative could be gradient ultracentriguation).
If you are choosing the single good 2D class, then the orientation distribution is probably quite limited. The symmetry is ameliorating the situation somewhat like you said. I suggest using 2D only to discard obvious junk and misspicks (see 4).
I see what @vincent is saying about the particle centering. I recommend template picking your low-resolution structure, it appears well centered.
Given 2), I think earlier 3D classification (heterogeneous perhaps using some of the bad ab initio classes as decoys) could help a lot, you may have a lot of empty SMALP nanoparticles (highly negatively charged and sticky) or other weird particles that are hard to segregate from unique views of good particles by 2D classification.
No need to apologize! I’m trying to squeeze all that I can out of this dataset since we’re still stuck at 50% capacity for the foreseeable future.
This protein does use undecaprenol phosphate as a substrate, I’ve been planning on supplementing my purified particles with it and collecting some additional data, I could easily add a superose column into the workflow prior to preparing grids.