Hello,
I am trying to solve a complex structure of a protein complex. Protein A is an octamer and the total MW is around 800 kDa. Protein B is a hexamer ATPase (~180 kDa in total) and binds to protein A when activated by ATP.
I collected a couple of datasets using different strategies to reconstruct the complex but got confused about where I did wrong…The target is to get like different intermediate states during ATPase turnover, but till now, only one dataset gave me resonable reconstruction.
In one of the datasets, I got like 30k particles in final reconstruction out of 1 million particles (6k micrographs). Some of the typical 2D classes after final refinement↓

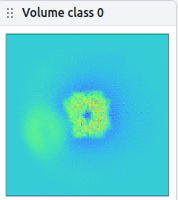
In another dataset, 2D classification in a mask diameter of the whole complex seems to be unable to distinguish A+B from just adjacent A+A (unfortunately, particles seem to be clustered even I tried lower protein conc. ). So I just excluded trash classes in the mask diameter of A and move onto hetero-refine (like what I did in the first dataset). But, in hetero-refine in C1, the refinement seems to be centered on A and only smeared density around it was observed. I am not sure, but is this something driven by the symmetry of A and low population of complex particles?
I found several similar topics here, and tried like symmetry expansion→3D classification w/o alignment in cryoSPARC, but it didn’t work… I’m considering to try with Relion symmetry relaxation though.
Do you have any advice on case like this?
Many thanks!