Hello,
I am working on a large-ish RNA-protein complex with one protein bound with some conformational heterogeneity.
3DVA followed by clustering has been pretty awesome at yielding a discrete conformation.
The cluster structure is binned (256 from 512), so I scale back up to full size particles using NU-refinement (which seemed to work better than homogeneous refinement).
As I suspect the static part of the complex dominates refinement, I tried to follow the NU-refinement with a local refinement, masking around the protein of interest (with no particle subtraction for now).
However, this led to an overall loss of resolution (as I understand things, that would be likely, as the resolution of the NU is at least partially influenced by the static part), and, more importantly, the map looks worse (see attached).
- Is there something inherently wrong using local refinement after NU-refinement?
- Which of the following are sensible things to try:
adjust the mask to include a larger area around my region of interest (making it “easier” for the program to align)?
adjust the fulcrum of the mask (here it was centered on the mask, which I would not consider too bad)?
adjust the settings of local refinement (likely reducing the angle/shift values, as the NU-map was not too bad)?
Or maybe all of the above?
Thank you very much!
Kind regards,
Luca
1 Like
Hi Luca,
How are you scaling back up to full size particles using NU-refine - what exactly are you doing?
There is nothing wrong with using local refinement after NU-refine. If you found NU-refine beneficial, you may want to switch on non uniform regularization in local refinement as well, if it isn’t already on.
Cheers
Oli
Hi Oli,
the input for the NU are the particles and volume from the cluster that looks good, then I pull the blobs from the unbinned original refinement (the one that was used for binning).
Is there a more correct way of unbinning?
In the local refinement, non-uniform refine is enabled by default (and in my jobs).
Kind regards,
Luca
no, that should work… how big is the region you are masking?
Around 130 kDa (protein + RNA binding site).
The complex is ~1.5 MDa (ribosomal subunit).
I am testing particle subtraction, but so far that has not really improved the situation.
Maybe I have to test a number of different masks for particle subtraction to find the optimum?
To go back to my original question: are none of the settings in local refinement suitable for “constraining” the refinement to the search space around what the NU-refinement yields?
To my unexperienced eyes it just seems the local refinement is “losing it”.
Also, is there a benefit to using a mask for the NU-refinement?
Kind regards,
Luca
Dear @cryo-lg,
Does the mask used for local refinement have a soft edge? If not, you can use the Volume Tools job to pad the mask to give it a soft edge, which is necessary for small masks.
Other than that, if the mask is quite small, there might not be enough information in the data to align, and your intuition is right that local refinement needs more help constraining the search space around the original poses. We added a simple feature to do this regularization – there is a parameter to activate a gaussian prior over rotation and shift, where the prior is centered around the original poses (unless the recenter shifts/poses parameters are on).
It should be one of the first parameters under “Alignment Parameters”, called “Use pose/shift gaussian prior during alignment”. You can specify the standard deviation of the prior over rotation and shift below that – generally it’s important to tune these values to the size of movement you expect to see. If you have a complex that undergoes a large amount of motion (e.g. a spliceosome head region, for example), you might want a larger prior like 20º and 10Å. On the other hand, if you are using local refinement just to try to improve detail in a specific region of the map and don’t think there is much independent motion of that region, much smaller priors, even as small as 3º and 2Å may be necessary. This use case (small masks) is touching on the limit of how much information can be extracted from the data, so it is still a bit experimental and for regions lower than ~150 kDa it’s hard to predict how successful it may be.
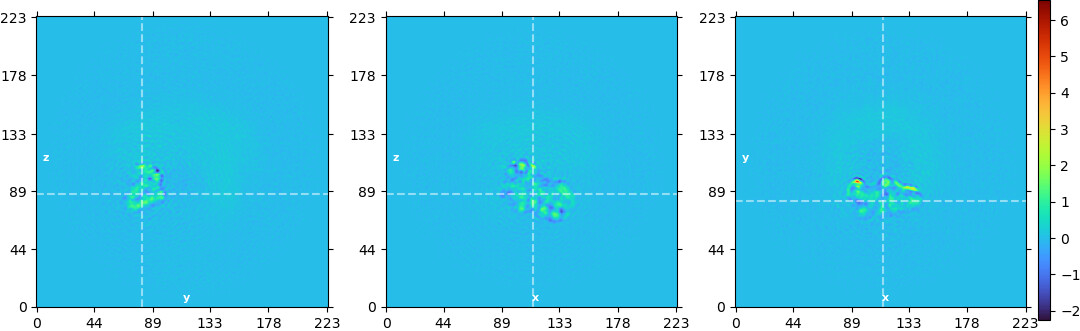
Edit: here’s an example of how the prior can help on an example dataset, the TRPV1 (EMPIAR 10059)
Here are the slice plots from a local refinement using a rotation standard deviation of 3º and shift deviation of 2Å:
And from a local refinement with no prior:
The prior was necessary here because the mask was very small. Without the added regularization, a lot of overfitting is visible and the map is uninterpretable.
Best,
Michael
4 Likes
Thank you!
I am going to try these things and get back to you.
I do treat all the volumes I generate by adding dilation (5) and padding (5), and setting the threshold to 0.05.
Is the padding value ok?
Kind regards,
Luca
Edit: just so I am not always complaining  the local refinement works great for larger masks in my structure, where there is a noticeable improvement in map quality.
the local refinement works great for larger masks in my structure, where there is a noticeable improvement in map quality.
Ok,
I have tried using the gaussian prior setting, and I have reduced the SD two 3° and 2 Å, but I am still losing definition. I am trying to incorporate particle subtraction now, maybe that will help combined with the rotation/shift constraints.
I was also thinking about changing the fulcrum to be on the edge of the small protein I want to refine, instead of in the middle.
Let me know if you have any other suggestion.
Kind regards,
Luca
Hi @cryo-lg,
This is probably entering the realm of speculation, but I’ve generally seen good results when the mask falloff is several factors larger than the resolution you expect to get to. Let’s say 5 times or so the resolution – so for e.g. if your dataset is getting to 3Å, and you have a 1Å pixel size, you want your mask falloff (the “Soft padding width”) to certainly be larger than 3 pixels, under this rule of thumb you might use 5 * 3Å / 1 Å = 15 pixels
Best,
Michael
1 Like
Hi Luca,
The only other thing that comes to mind is that you may have some compositional heterogeneity (presence/absence of the subunit of interest) in addition to conformational heterogeneity. In this case you might get some benefit from isolating a higher occupancy set of particles by performing masked classification without alignment in relion or similar.
Also what initial lowpass are you using in local refinement? For small volumes, going just a bit worse than the global refinement can be beneficial (e.g. 8Å filter for somewhere that previously had an estimated local resolution of 4.5)
Cheers
Oli
2 Likes
what Michael said too - also the smaller the mask, the softer the edge usually required to avoid the mask edge effects that can cause problems with convergence to high resolution.
Hello and thanks for the feedback.
I was definitely using a smaller padding.
This will also address Oli’s point:
I start with an ab initio, followed by heterogeneous refinement (8 classes), where I remove the “most” apo class (i.e. no density for my area of interest even at very low threshold).
I then did two local refinements on the ribosomal subunit to get the most definition out of that (~3.4 Å).
To look for my protein of interest, I pooled the 7 classes into a 3DVA with different masks, with the visualization using clustering to generate discrete volumes.
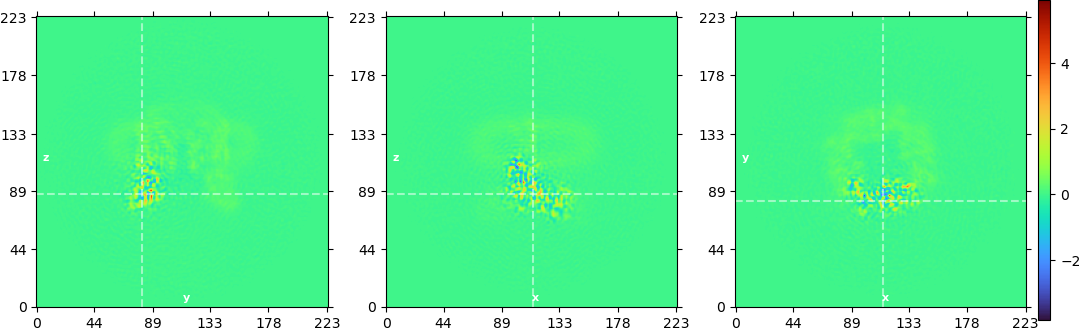
@olibclarke : I am from here on only focusing on particles from the stably occupied cluster (left image in the top figure).
That’s when it gets bumpy: the NU-refinement reaches ~3.9 Å (my protein of interest has a worse local resolution, but still ok-ish - that’s the middle image of the figure).
At this point, local refinement around my protein of interest is causing the problems I mentioned (as shown in the figure on the right side - all density of my protein of interest just looks worse), and resolution is stuck at 5.8 Å.
I will try increasing the mask (both absolutely, by adding neighboring density around my POI, and through the padding as Michael suggested).
The current refinement is still running, but I am seeing a small benefit of using the subtracted particles (the same job that before was at 5.8 after 4 iterations, is now at 5.65 at iteration 2).
@mmclean: if I understand you correctly, you are saying the padding parameter overlaps with the dilation, so if I am using 5 and 5 for dilation and padding, I have 0 mask falloff.
Kind regards,
Luca
Ah shoot my mistake, the original message was wrong – it should just say “Soft padding width” and not the difference between them. So if you use 5 for both parameter, the mask will have a falloff of 5.
Hi Luca - you may find that a masked classification without alignments does a better job of separating your apo from your holo classes (perhaps as a second step after heterogeneous refinement and an initial NU-refine step)
Hi Oli,
I thought that’s kind of what I am doing with 3DVA clustering.
Also, do you mean in relion or is there a feature now in cryosparc?
Kind regards,
Luca
3D-VA clustering is very different. Yes, I mean in relion (or other packages, but I am most familiar with relion), that feature is not yet available in cryoSPARC.
1 Like
Thank you, I will try that.
Kind regards,
Luca
Quick update:
as @mmclean suggested, I tried increasing the soft padding of my mask (from dilation 5, padding 5, to dilation 10, padding 25): great success!
Resolution is 5.8 Å, but it is pretty clean (before it supposedly was 5.6, but not really interpretable) . I can trace the whole backbone of the protein-of-interest, and I see blobs for larger residues that help adjusting the crystallographic model I am using.
This was done using the minimal map just around the POI, so I will now try to increase map size a bit to include the surrounding ribosomal proteins and RNA.
I’ll follow up once I have more updates (also with respect to @olibclarke suggestion regarding relion).
Thank you very much for the help!
Kind regards,
Luca
6 Likes