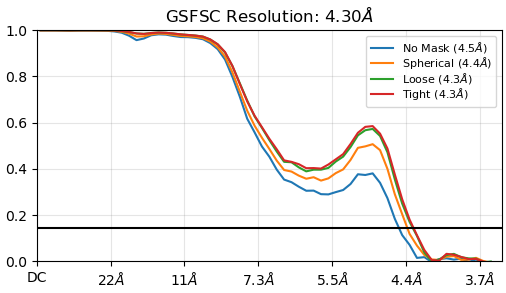
Hello, I was doing refinement of a small membrane protein(~80kD). I successfully got the outward conformation of this protein with 3.4 angstrom. However, when I looked into 2D class clearly, I believed there exists another conformation. So I used the same family of this protein which is inward state to isolate inward open particles. And I got 500k particles. After ab initio, whether I did local refinement or NU refine of initial model, I saw a clear shake of FSC curve:
But the density map looks not bad
With so many particles remained, I believe the final resolution could be 3.7 angstrom at least. However, this inward map can not successfully isolate inward particle when doing hetero refine with outward map and bad map as input. Besides that, the. NU and local can not improve resolution with first round is 4.5A and final is about 4.3A. When I did 3D classification, things got worse: FSC curve not correct and the map is distorted:
How can I do to get high resolution of this inward state?