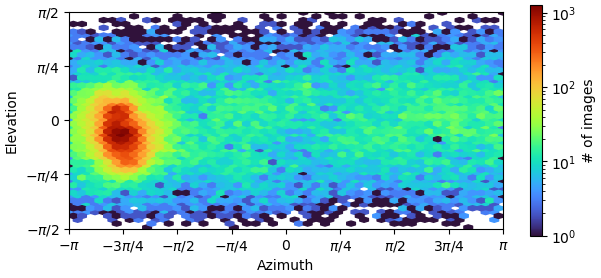
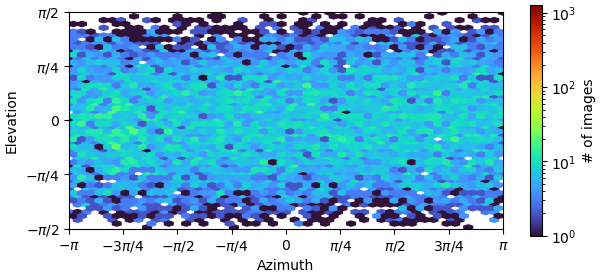
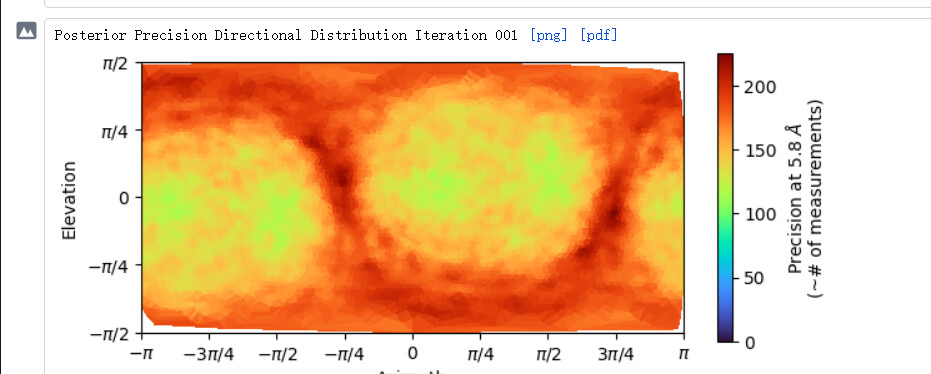
Hi. Recently, I was processing data o fa small protein, whose molecular weight is about 120kDa, for many rounds of refinement, but the problem of orientation advantage still exists. Strategies that have been tried so far are:
Manually do not select the dominant orientation in 2D classification for subsequent processing.
Perform rebalance orientation after Nu-refinement.
But the problem of orientation advantage still exists. Do you have any good solutions?Thanks.
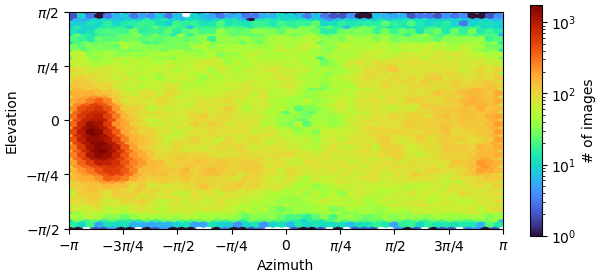
that is a pretty bad example of the issue. I strongly suggest using view_select to encompass several distinct regions that are NOT the dominant view (even though there are no obvious “hotspots”), running 2D classification with ~10-30 classes for each subset, then training a topaz picker for any 2D that looks different from preferred view. Simultaneously you should consider other ways to prepare grids (LEA? grid type/coating? detergents?) as this might not be insurmountable. In future if you have prepared multiple grids and you can manage to identify strong preferential orientation early, consider screening another - sometime’s the strong bias is grid-specific as it happens very quickly post-blot and is dependent on ice thickness and time.
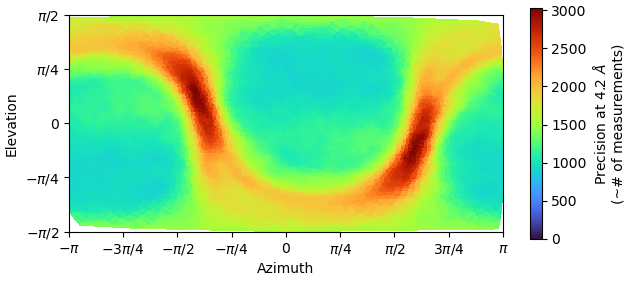
Often, you need to adjust the threshold for removal for optimal results. If it’s not helping at all, given the distribution of particles in that plot, I would run a quick 2D to check for junk.
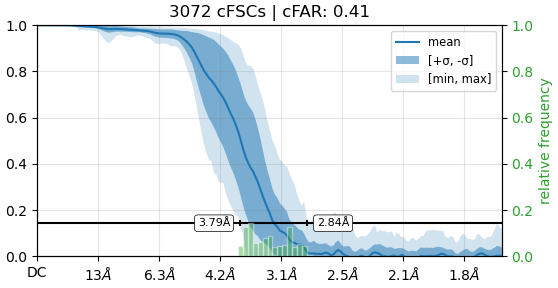
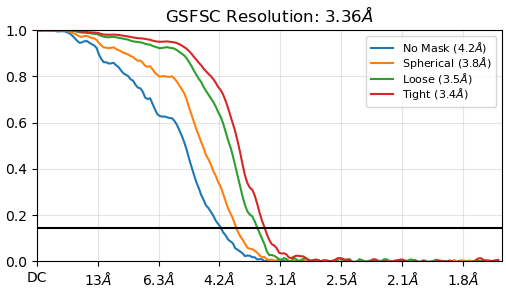
From the result of rebalance orientation,this seemed to be an improvement, but after running Nu_refine, I found that the resolution and cloud images were worse.
you desperately need more particles picked from a different view. I would revisit picking strategy and try to find any unique particles and use Topaz to select for more of them, iterating a few times to continue getting more and more