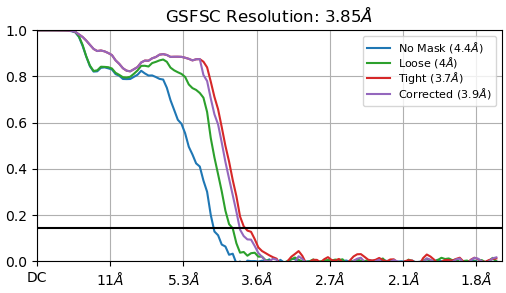
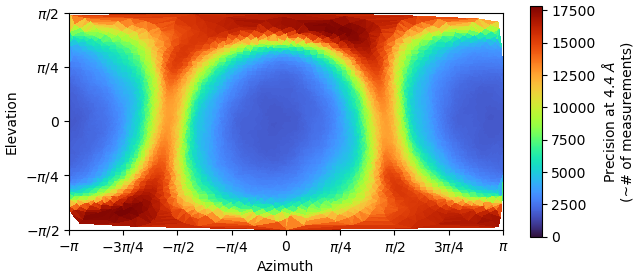
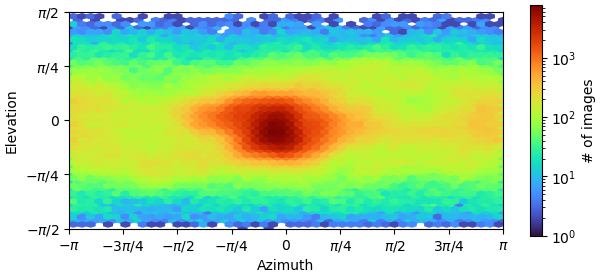
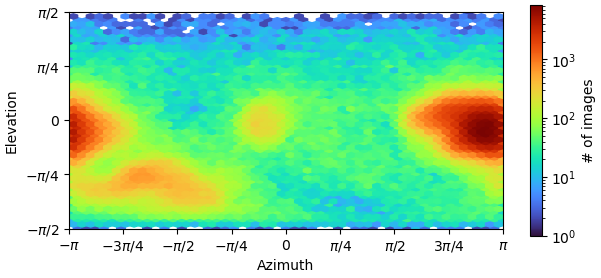
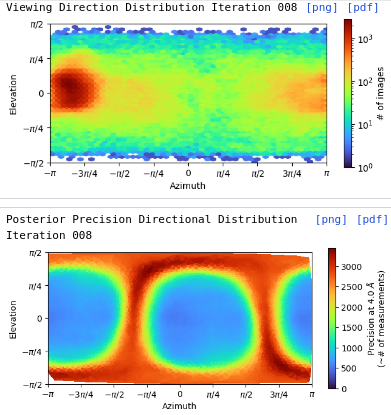
I am dealing with beautiful 2D classes, however from Direction Distribution it looks I have preferred orientation. I have more that 700k particles, with obvious nice secondary structures so it is quit frustrating . What do you think? How can I improve resolution, now I am at 3.85 and I am not able to push it down. Any tips for some better 2D classification to avoid rare views loss? I’ve tried 3D (all classes looked similar), heterogen. refin, NU refin. Or should I collect new dataset?
Check what the 3D looks like on that reconstruction because I’ve seen strong preferred orientation give high resolution fscs, but the map is completely useless…
Otherwise, clean obsessively, pick all classes except the dominant view, se where you get. While the dominant view is obvious, there seems to be a reasonable scattering of other angles if the distribution is to be believed…
700 K particles @AlzbetaD assuming orientation bias ? Maybe do a hardcore 2D classification job (140-200 classes, 40 online EM iterations, etc) => select 2D see if there is any obvious junk => put these in the Rebalance 2D Job (Down sampling factor =4, rebalancing=1, number of super classes = 6-10) => see how many particles get through.
Topaz or CrYOLO will help to pick unbiased. Sometimes with blob=>templates the humans are bias. If you do get 2D classes if the rare views you can go back and re-train TOPAZ with a better set of particles.
This does not look too horrible to me. I would advise trying the opposite: don’t do even more 2d classification, but go back to the start and skip it entirely instead. You might lose rare views in extended runs of 2d as they don’t align well. So,
Take all your extracted particles and put it into heterogeneous refinement with 1 good reference (your 3.85 Å reconstruction) and 5 bad references (many ways of getting these. you could run ab initio on 1000 particles you excluded in your first Select2D job, for example). You can decrease refinement box size to 72 or 96 to speed up the process
Repeat this for 3 rounds, only keeping particles from the good class as input into the next round
Run homogeneous refinement and compare particle number & viewing distribution with your current ‘best’ map. Do you see better defined features? Less stretchiness? Better viewing distribution?
If you have more particles now, maybe that already took care of your problem. If it did not help, but you do have more particles, i would try classification again, basically just like you already did (but this time with more particles). Sometimes 3DClass helps (‘force hard classification’ on, otherwise I never get good separation), sometimes Heterogeneous refinement with 8-16 good classes as reference (sometimes magically one class with completely balanced views appears) works. If non of that works, back to the drawing board and screen detergents or think about data collection on a tilted grid.
NB: I always skip 2d nowadays (i have 2d running in cryoSPARC live, of course, as a monitoring tool), and it helps in probably 60% of cases and yields same results as ‘with 2d’ in 39%. The only times it fails is if you have very strong artifact-features in your particles (e.g. picks on carbon edge or beam ripples), as these particles tend to get smeared into your nice protein class. In these cases a single round of 2d only to exclude artifacts helps.
I’ll second @Moritz here, I’ve dealt with this issue you’re having too. If you do 2D at all, just do a very gentle cleanup where you get rid of the most obvious junk (edges, large bright spots). Then use multiple iterations of Heterogeneous Refinement where particles from good classes are classified against a good ref and multiple junk refs for more complete cleanup. This is more robust to preserving rare views than 2D selection.
There are a few threads on this forum that discuss this approach, sorry I don’t have them at my fingertips though.
Side notes: Many rounds of 2D can still be useful to get ultra-clean subsets for Ab Initio references. Stopping an Ab Initio run early can be a good source of junk references.
Also, if your map is pretty good and still has some streaky features from this preferred orientation, sharpening with EMReady or DeepEMhancer can help.
I’ll also second @Mark-A-Nakasone, I’m a big fan of crYOLO, even just with the default model, to pick particles without as much junk and more rare views than a blob picker.
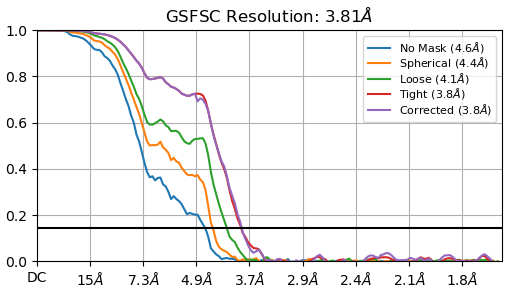
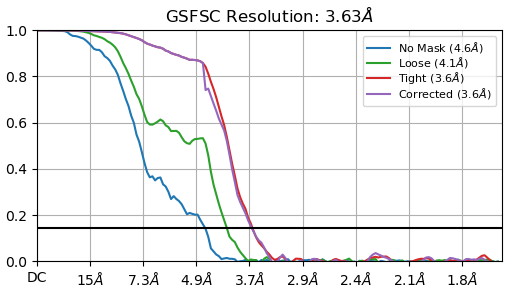
Thank you all for your advices! I will apply them and we will see…for now I have tried multiple heterogenous refin., bigger extraction size, and multiple 2D classifications. I think that distribution is better and for now resolution is from 3,6-3,8 A (for homogenous refi) but I do not like in general my curves (added). Do you think is better o use NU instead of homogenous refinement? Additionally, in NU is B-factor quite high, 190.
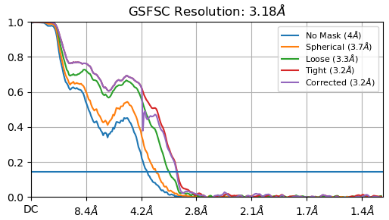
I am facing a similar dilemma. I have more than 300k particles and the resolution as per the FSC appears to be 3.2, I am concerned that there is possible preferred orientation that I am dealing with. I have attached the FSC and distribution plots below. Your suggestions on how to get over this problem through different processing strategies will be much appreciated.
Maybe a bit late to reply, but I recently observed that combining extensive automatic orientation rebalancing (using the new ‘Rebalance Orientations’ job in cryoSPARC v4.5+ and playing with the ‘rebalance percentile’ parameter) followed by 3D refinement with Blush Regularization in RELION v5 can result in perfectly interpretable maps from datasets with extreme preferred orientations.
Hope this might help!
Sounds exciting - can you explain a little more here - how are you tuning the “rebalance percentile” parameter, any typical values? Do you rebalance orientations using the orientations from Blush or CS? Also what exclusion criterion do you use for rebalancing? Random or 3D-error etc?
I’m also interested in learning what rebalance orientations parameters may be helpful.
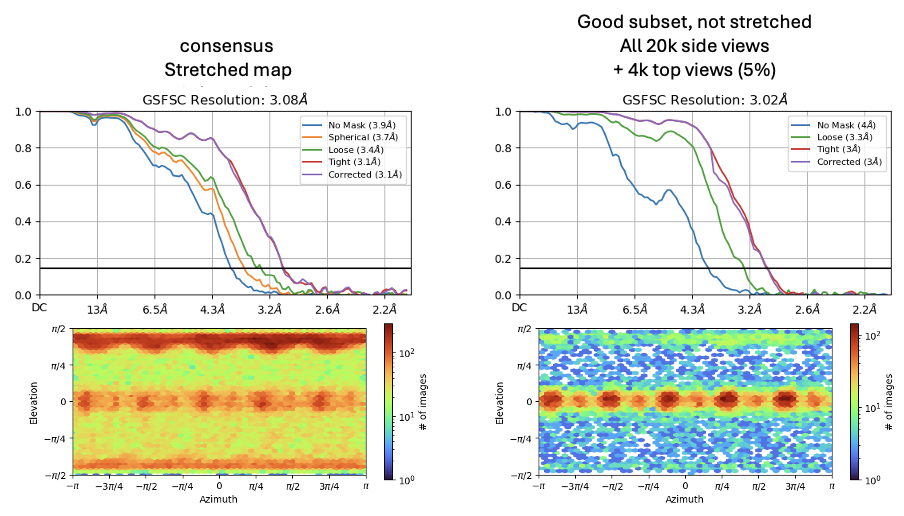
I have a dataset with predominating top views that gives a horribly stretched map. Rebalance orientations hasn’t given a nice result in improving the map quality. Manually removing 95% of the top views (select top views by 2D and remove with particle sets tool) yields a nice map. I’ve not been able to get nearly as good a result using rebalance orientations. This is a C5 symmetric protein, so a single side view perpendicular to the symmetry axis gives 5x views of each protomer.
The following Rebalance Orientations parameters yield 6-8 Å maps of poor quality. It can’t seem to find the good side views.
In my particular case I used Orientation Rebalancing after NU-refinement in CS. I did two rounds of Orientation Rebalancing (with random selection of particles), first using a percentile threshold of 60 and the second time a threshold of 5. After the first round of rebalancing I redid Ab Initio reconstruction followed by heterorefinement and NU-refinement of the best (and most isotropic) class.
The percentile thresholds were chosen empirically by checking Orientation Diagnostics jobs and observing both obtained resolution and improvements in cFAR scores. I then used 3D refinement with Blush regularization to get rid of streaky artifacts that were still present in the final NU-refinement after two rounds of Orientation Rebalancing. cFAR values significantly improved after performing the final 3D refinement with Blush Regularization.
Hope this is clear,
Best, Jan
For another case I recently worked on (where I had a non-tilted and 40 ° tilted dataset) I also observed that removing literally all of the non-tilted data (which only had top views) was necessary to get isotropic maps.
For the particular case I describe above (see answer to Oli) there were only top views to start with, no side views to be seen at all. Only after Orientation Rebalancing + Blush I could observe new tilted views that appeared in a 2D classification of the final particle set.