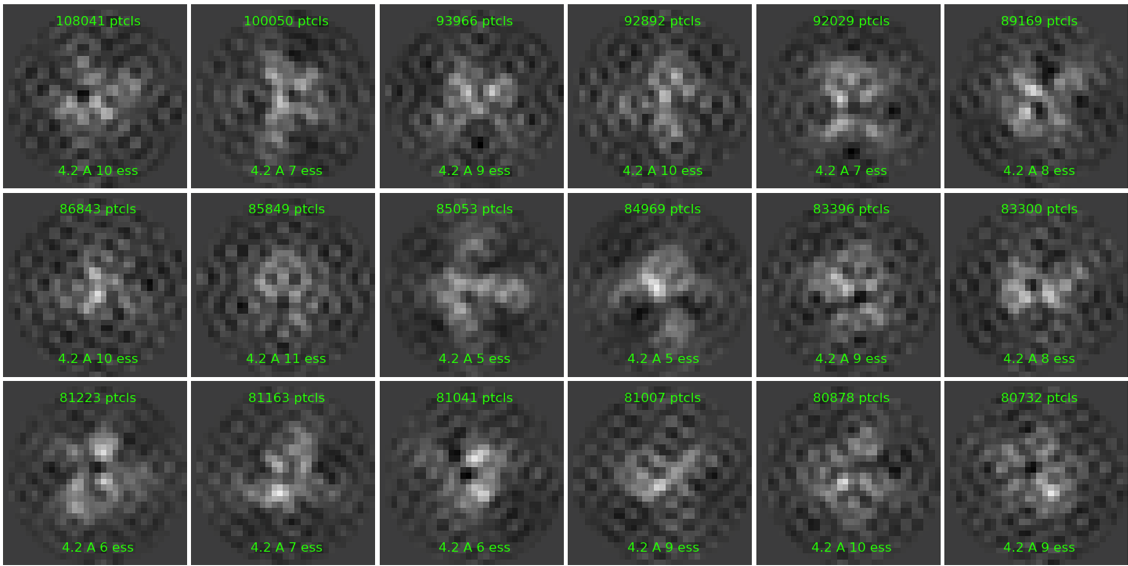
I am trying to work with a very small protein for the first time and I am confused with the results on the 2D classification since they look pixelated and I have never seen this before. The pixel size is 0.73 A/pix and I performed Patch Motion Corr and Patch CTF. I followed by curating the exposure and only taking the ones with a resolution estimation between 1.7 and 3, I also discarded any outliers on defocus, average intensity and so on. I ended with 4623 micrographs and did blob picker and extraction on two box sizes, 84 px and 140 px always around 6.5 million particles. I have tried doing 2D classification changing the parameters online-EM iteration to 40 and Batchsize to 400. In every case I end up having pixelated 2D classes, which I am not sure why it happens. Any idea why I may get this? particularly since I do not do any binning of micrographs or particles at any given time.
I would understand if I do some binning that lowers the resolution to that extend, but I do not perform binning and I was following a similar publication processing. Any thoughts?
It’s overfitting. Looks like it’s aligning noise in the particle boxes because the actual protein signal is too variable for class assignment.
How many classes do you set? 6.5M particles into 50 classes will not go well, especially since they look quite heterogeneous.
For 6.5M particles, try 300-400 classes or so (and 200-400 particles per class per batch).
Also it would be worth looking over your picking very carefully and checking that you can (a) see particles, (b) they are well centred in the picks and (c) that you don’t have empty ice picked (more than necessary).
There are a lot of possible options to tweak, but the above is probably a reasonable starting point.
Another important thing: your pixel size is much finer than it needs to be for 2D classification. In the extraction job, use the option to Fourier crop your particle images such that the final pixel size ends up in the ballpark of 3-4 Å/pix.
This will have two beneficial effects:
a much smaller box size (in pixels; but same physical size in Å of course), so much faster computations (the time it takes to compute scales with the total number of pixels, so with the square of the box size!);
coarser pixels will limit your attainable resolution, but for 2D classification this is actually beneficial because it flattens the noise and because 2D alignment and class assignment only need medium to low resolution features; I think it will be helpful in your case, with the alignment being seemingly significantly driven by noise.
And one more piece of advice when you have so many particles: if you want to experiment quickly to figure out good parameters for the 2D classification job, use the particle set job to make a random subset of only a few percents of your particles. Make sure you activate the random option, so this subset will be representative of the whole set. Then you can try testing parameters with much more rapid feedback than when running the job on the entire dataset, and once you have a set of parameters giving good results you can run a job with all particles.
I’ve found for some really small samples, Fourier cropping the particles causes more problems. It’s worth trying, of course, but small proteins (especially in strong detergent densities) have enough trouble aligning well already, heavy binning can make it worse. Furthest I would take a small complex would be around 2Å (so a 3-fold crop might be OK).
It looks like you picked too much junk. Your 2D averages are overlaid on what appears like averages of noise. Try several rounds of 2D with lot more classes, as suggested above. Then train Topaz on good subset.
Also, cryolo with general model might do a much better job at the initial picking.
Piggy backing off of some of the other user comments here as they have offered good advice, you might see good results by doing some of the following:
Cropping particles during extraction. I would recommend something like 180 or 256 px extraction downsampled to 60 or 90 px, respectively. This will help with speed and wont limit the alignment resolution.
Next, use more than 200 classes or maybe split off a subset of particles (maybe 1M) and perform 2D classification on those, train a TOPAZ or crYOLO model. Using a NN picker, especially for small proteins/complexes, usually provides better results for limiting the number of junk pics and picking of more rare views.
The batch size and # of online-EM iterations looks appropriate.
Lastly, for small particles, turning off Force max over poses/shifts might give better results and help to eliminate the pixelated features you are seeing.