Hi all!
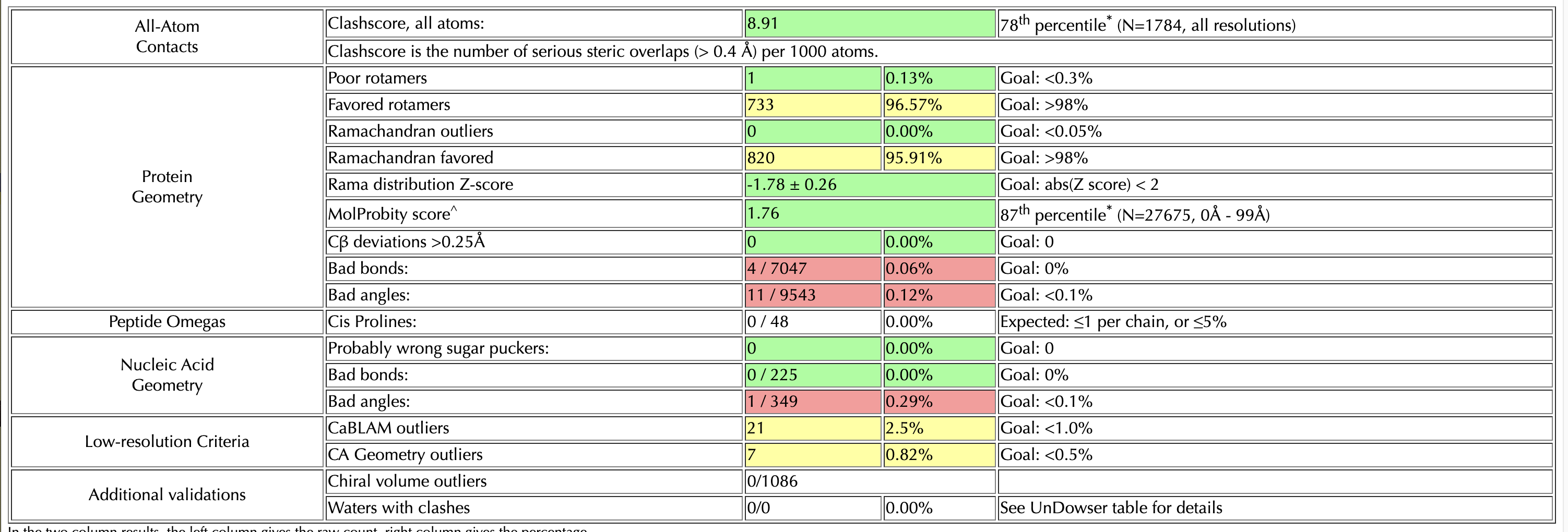
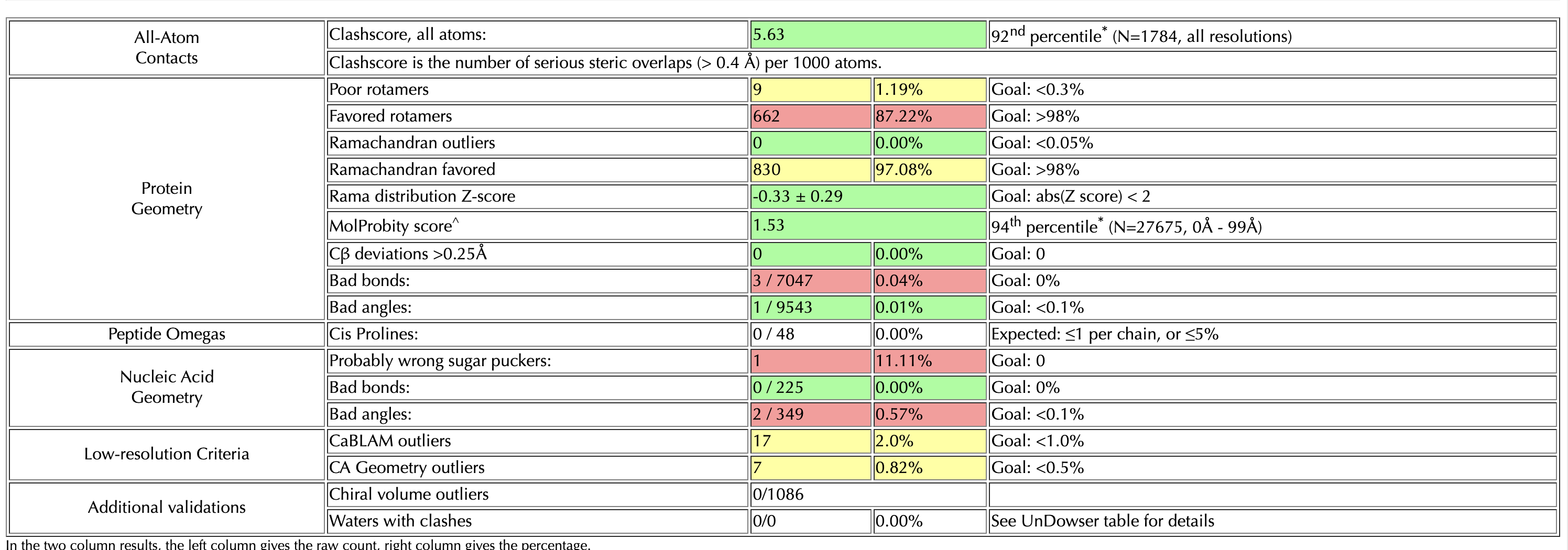
I’m doing rounds of Coot and Phenix real-space-refine on a 3.2 Angstrom map. I’ve reached a point where my model looks good after Coot, when I check it on the MolProbity server. However, if I put it into real-space-refine, the statistics get worse – generally, the bond angles and lengths improve (I have ~10 outliers in my build) but the rotamers get worse. I’ve tried without local grid search, and with the ‘rotamers’ box unchecked - but this made my Ramachandran get worse. I’ve also gone through the build res-by-res and optimized rotamers for the map, while keeping an eye on Ramachandran plot (which was good after Coot). I’ve attached my two molprobity statistics pages for reference (top one is my Coot build, bottom one is a phenix output with default settings)
I have two major questions:
- Is it considered bad practice to submit a structure that is straight out of Coot, provided the statistics are good?
- How would I combat the problems that I’m getting from Phenix?
Thanks for any advice!
-Leah
Hi Leah,
Regarding question 1, if you are sure you prefer the coordinates from your manual fitting, then I suggest running phenix.real_space_refine with everything turned off except for B-factor refinement, because you still want them correctly refined even if you don’t want the coordinates to change. I don’t remember if coordinate refinement can be completely turned off, but if not you can set reference-model restraints with your input model as reference model, and set the restraints as tight as you can make them: this should equivalently prevent coordinates from moving much or at all.
Regarding question 2: maybe try a different refinement program? I like servalcat these days (it’s working in reciprocal space though).
I hope this helps!
suggests a minor deviation from correct pixel size. it’s crunching or expanding the model to fit a slightly too-small or too-large map. is there a xtal structure available, and can you overlay/interpolate/look for “breathing” motion large to small. Can DeepEM different pixel values (0.05A steps) and run map-to-model correlation for known crystal structure that is fitted to each resulting map and plot the CC values to find a global minimum.
1 Like
Hi Guillaume,
This makes sense, thank you! I wondered about using the same model file as input and reference, whether it would confuse the program or not. Good to know this can be done.
I’ll have a look at servalcat, thank you!
Hi CryoEM2 - this is interesting. Thank you. What can be done in the case of no crystal structure? Also, where is the discrepancy in pixel size? (ie, am I processing in Cryosparc with a different pixel size compared with the collected data? Or is the error introduced elsewhere?)
Thanks!
Leah
without crystal reference for this (or any other structures collected at this pixel on this microscope and solved to high res) you will need to have the microscope operator perform calibration and provide a slightly adjusted pixel size.
in this scenario, the full cryosparc processing was done at an incorrect value since import/live processing so the maps it produced all along are slightly the wrong size.
there’s a decent chance this is not your issue. could also be overfitting at the mask edge. when you coot-correct the RSR, what are you changing? could also run linear interpolation in chimera of the RSR model and the Coot model to understand.