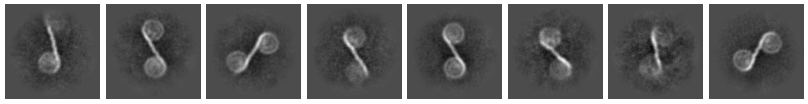
Hi,
I am processing a dataset with mononucleosomes in complex with my protein of interest. While processing, I could see classes with di-nucleosomes, that I would like to look into since my sample only had mono-nucleosomes.
I am planning to use template picker to pick more particles belonging to this class, but I am not sure what values would be best for the run.
These are the values for the job:
Particle diameter: 300A
Low pass filter (both template and micrograph): 20A
Angular sampling: 5 degrees
Use CTFs to filter the templates: ON
Min. separation dist (diameters): 0.5
Number of mics to process: Not set
Number of mics to plot: 10
Maximum number of local maxima to consider: 4000
I am also worried about not getting an accurate representation of the side views, since that would probably be hard to pick manually.
I would greatly appreciate any suggestions to improve my particle picking, and any advice regarding picking the side views.
Best way forward is to try your parameters on a subset of micrographs (~500-1000), perform 2D classification and see what results you get. Adjust values and repeat until you are happy, then you can pick from the entire dataset. Because every dataset is different, there are no default values that will always work for dinucleosomes but your starting values seem ok.
That being said, other particle pickers (Topaz, Cryolo) might give more clean results if your micrographs appear heterogeneous. You will have to try it out.
There is no recipe to obtain more sideviews, it’s unlikely they will be left out by any of the particle pickers. Once you reach the 3D stage you will have to assess if your reconstruction suffers from preferred orientations. If that’s the case you might have to go back to the sample or grid stage and change strategy (use detergent, different grid type, tilted data collection…).
Thank you.
I will go ahead and try what you suggested. I tried Topaz, but the extract job failed (AssertionError: Subprocess exited with status 1). I tried the extract job again with around 4000 micrographs because I had read in another discussion that it might help. But no luck.
I was also not sure what values for the ‘Min. separation dist (diameters)’ would work for the template picker job, because previously I used the default value, i.e 0.5.
for side views, which will look effectively the same for mono- or di-, you have to include them all and let something like heterogeneous refinement sort out which contain the depth of information to belong to dinucleosomes. You can also use “create templates” of a 3D model of what you think each of these volumes is going to look like, and select side views which are similar but distinct between the two models and use those to pick. But picking with these “created” templates doesn’t work very well in my hands.
Hi,
I will look into creating templates of the 3D model if the template picker doesn’t pick any side views. Thank you for that suggestion.
Previously, I did try inputting 2D classes of mono and di nucleosomes, both into heterogeneous refinement. But, the side views I had then had all been mono nucleosomes, and the 3D structure for the di-nucleosomes was essentially two spherical volumes. But I was only picking mononucleosomes during that processing. Hopefully, this time it will pick some side views of di-nucleosomes as well.
they will also be SIGNIFICANTLY rarer views due to the size more than doubling, so might have better success in thick ice, or modified grid (carbon/graphene), if you were really interested in the structure