Hi all,
I am new to cryoEM and cryoSPARC. I am working with a small protein complex (100 kDa) in my master thesis and I am having some problems with the 3D reconstruction. The dataset was collected with volta phase plates.
I have only about 70-100k particles.
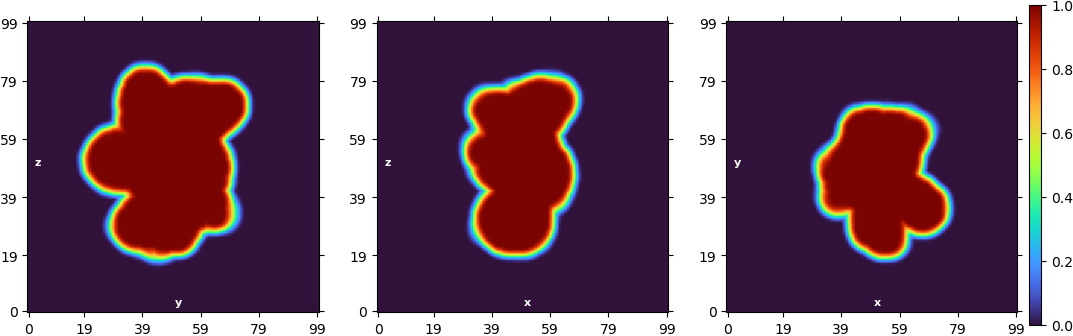
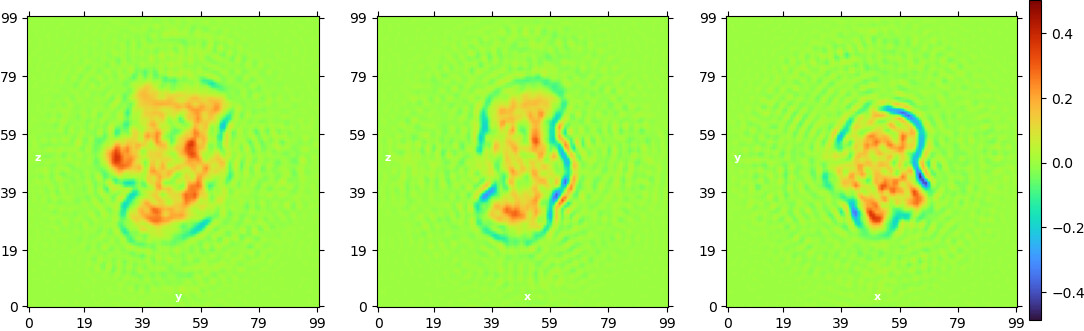
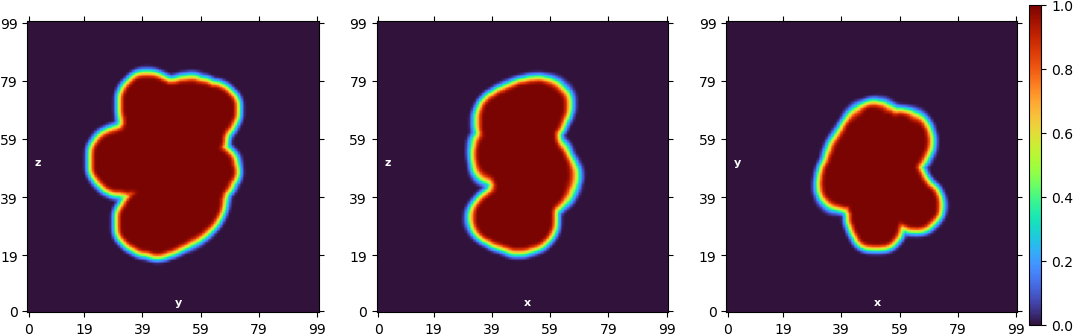
The ab initio models don’t look bad, but when I turn into the refinement, the “new” homogeneous refinement overrefines the model and “breaks it” completely. If I use the “legacy” algorithm or the non-uniform refinement, it also produces overfitting.
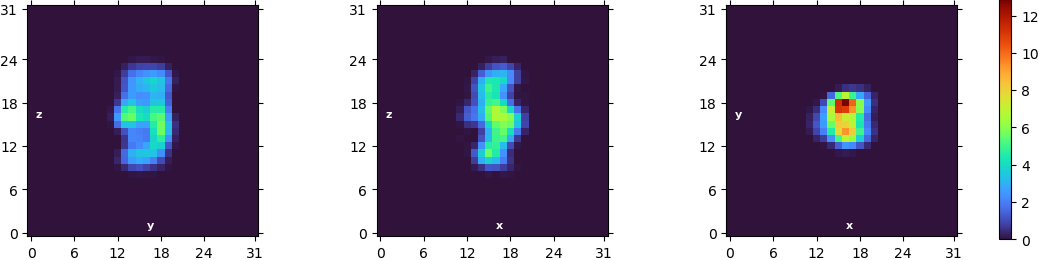
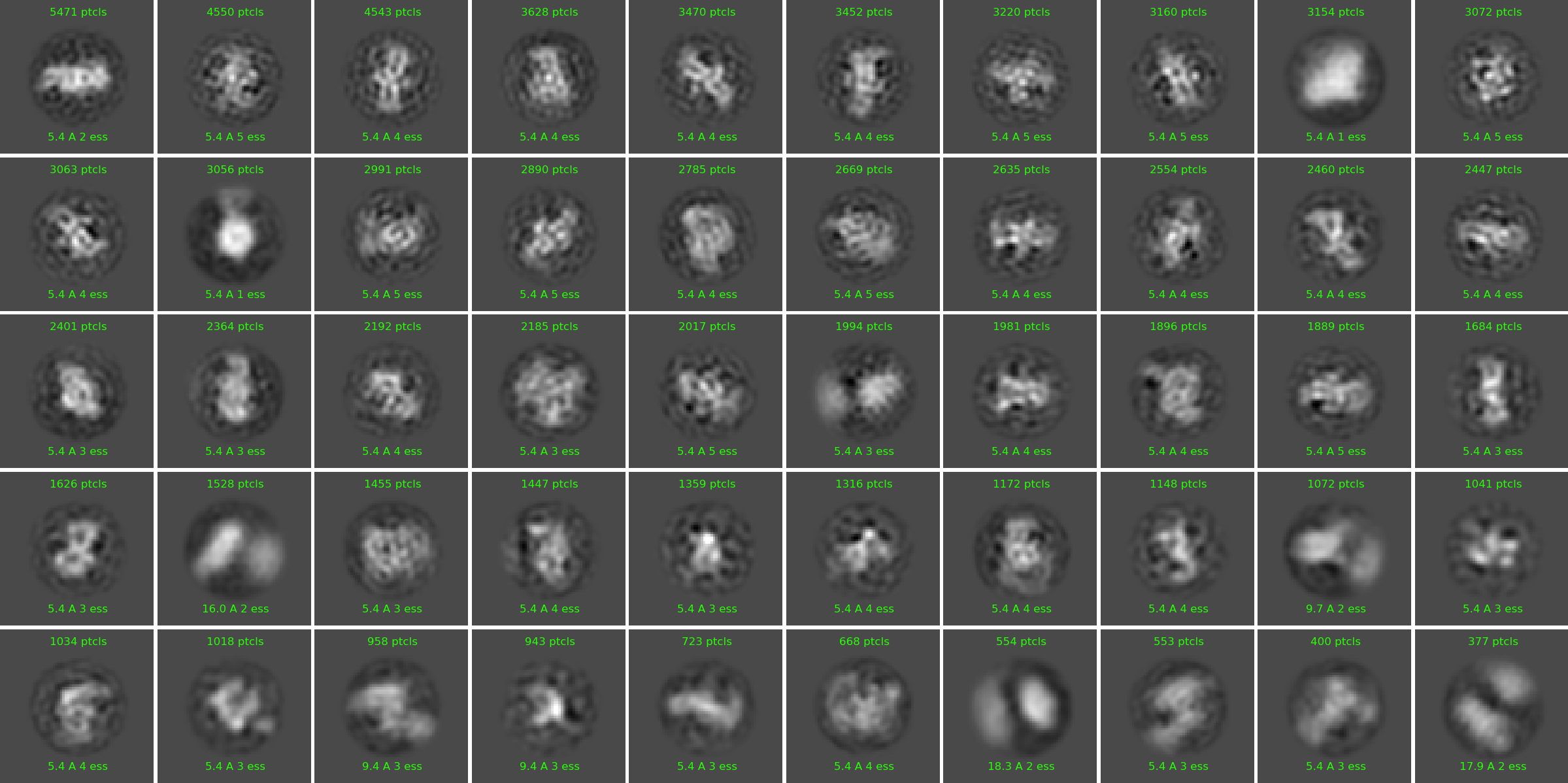
Example of ab initio model (71k particles):
“New” homogeneous refinement:
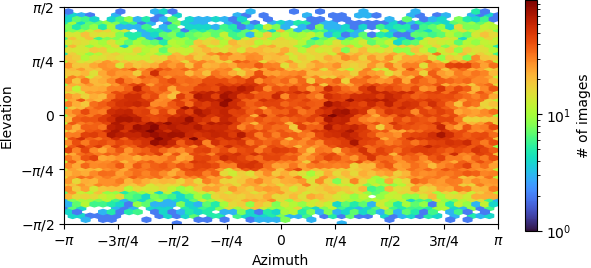
Non-uniform refinement “New”:
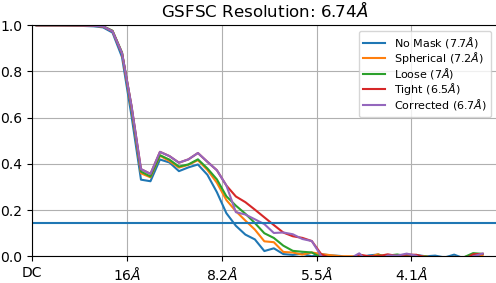
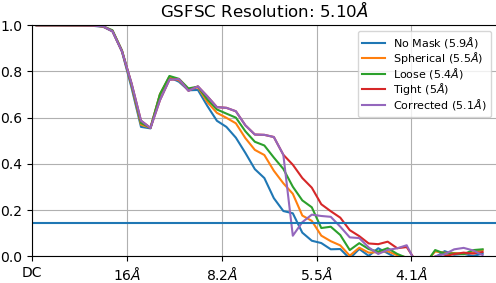
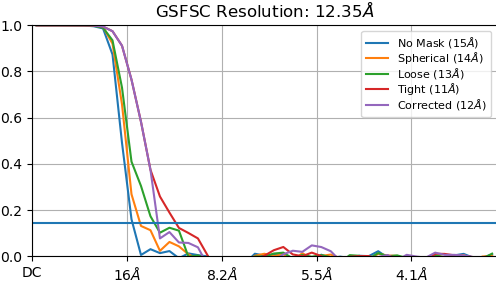
![P4_J1060_fsc_iteration_004_after_fsc_mask_auto_tightening|496x283]
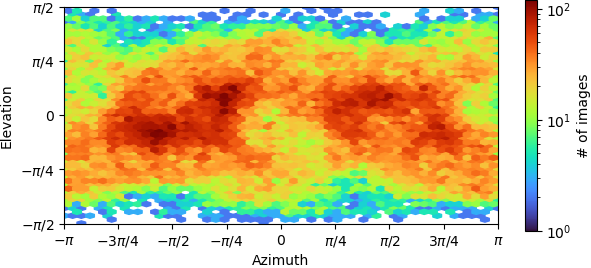
(upload://5imET93ikf1DFbR8KEVEcMR2Vp0.png)
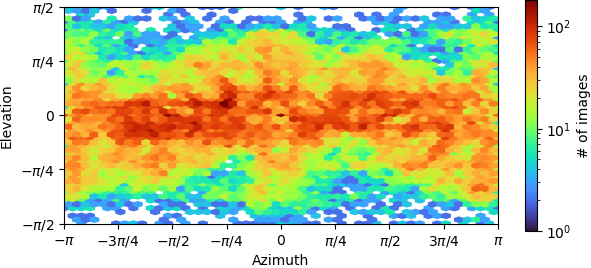
“Legacy” homogeneous refinement:
If tried tweaking the parameters of the dynamic mask to make it softer, but it has not helped. I also tried limiting the alignment resolution, performing multiple ab initio classes and doing heterogeneous refinement. But I always get overfitting. Is it that this dataset is not good enough or I do not have enough particles?
I should also say that the 2D classes look “grainy” unless I turn Force max over shifts/poses off.
Thank you!
I don’t have a direct answer to your questions, but here are my observations:
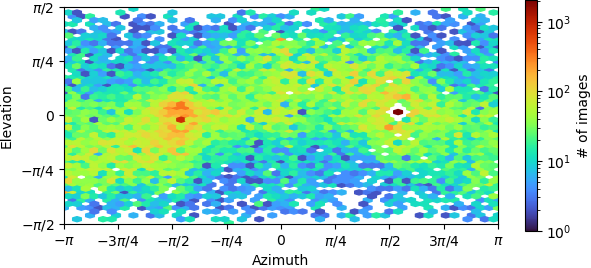
- The elevation/azimuth plots of the ab initio refinement indicate you have a restricted orientation at (±pi,0), which may be problematic. You can sometimes overcome this if you collect enough data and then discard a large fraction of the particles in those views, so that the angular distribution is more uniform.
- Your FSC plots indicate you collected this data at (or down-sampled it to) 1.6 Å/pixel. I suspect this may prove too course for a small particle like this. You’d probably do better to take the micrographs at 2x the mag, which requires collecting 4x as many images. Yes, it’s a trade off.
- I’ve no experience with Volta plate data, but when I run into problems I always want to check whether the CTF correction is right, and whether the particle picking is decent (i.e. not a lot of background picks). I assume you did 2D classification to get rid of junk particle picks?
Best,
RJ
Thank you for your answer!
Yes, the data was collected at 0.82 A/pix, I down sampled to 1.6 A/pix. I removed junk particles with 2D classification, but still the classes look “grainy” (overfitted but not with streaks), unless I turn off Force max over poses/shifts.
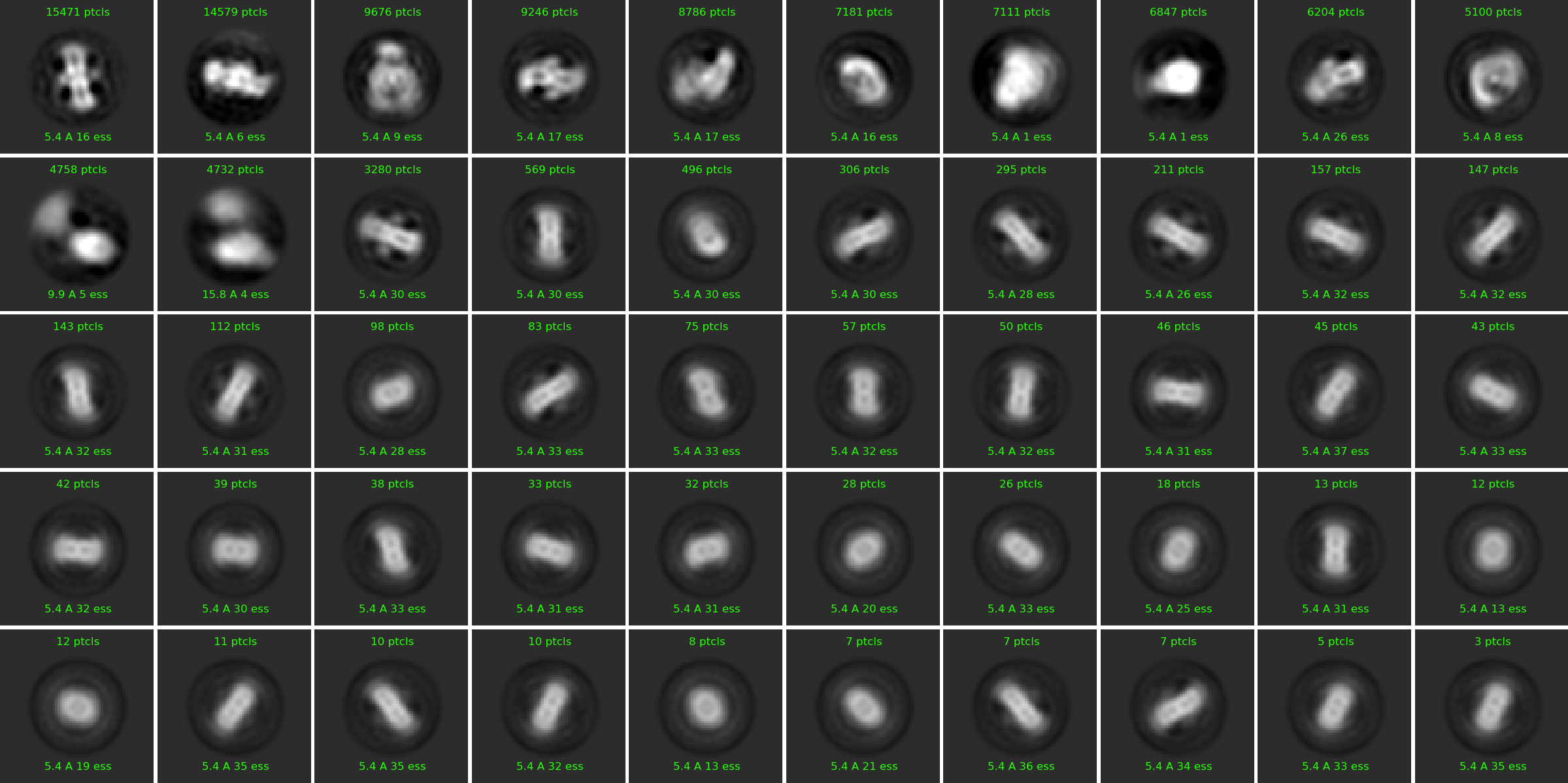
This is one example of one of the last 2D classification:
And this is if I turn off the Force max over poses/shifts:
The final classes I retained were these:
I think the CTF estimation is not that bad, with an average fit of about 4-5A.
Thank you again for your answer!
1 Like
Have you had any luck with this problem?
I have a similar problem, FSC drops very quickly to ~0.5 at 12A and then drops below threshold at 4A. The maps look like the particle but no refinement or details so to speak of.
Hi,
Unfortunately no. We decided to collect new data and I am currently working on that.
Good luck!
Hi,
I am also working on ~100 kd small protein and saw something similar, just wonder what your raw image look like? What concentration you used to freeze the grids?
Hi,
I used around 2 uM and that seems like a good concentration.