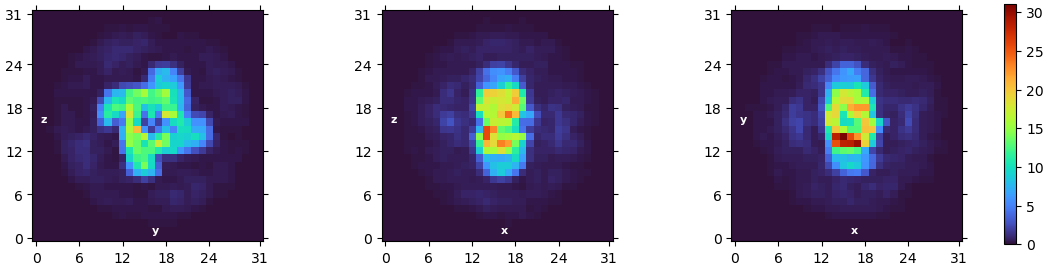
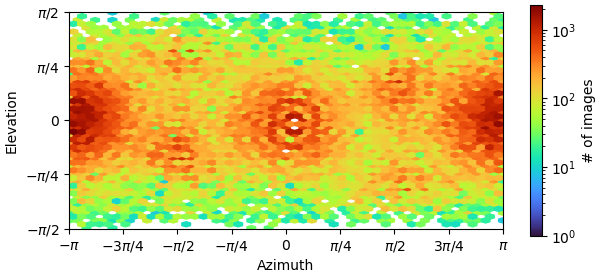
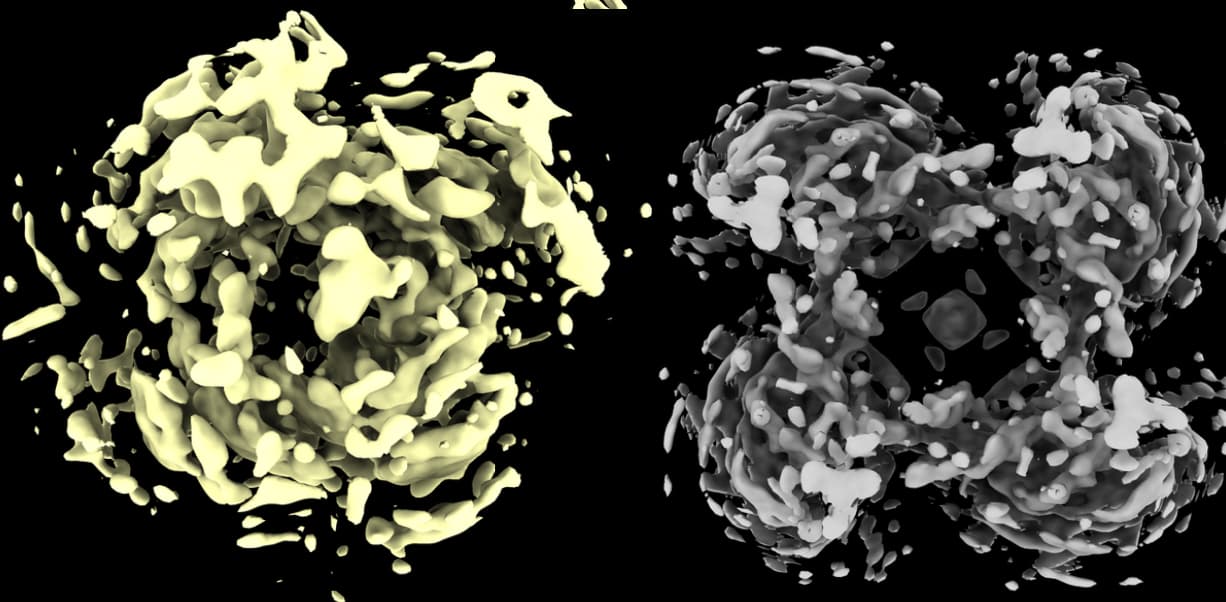
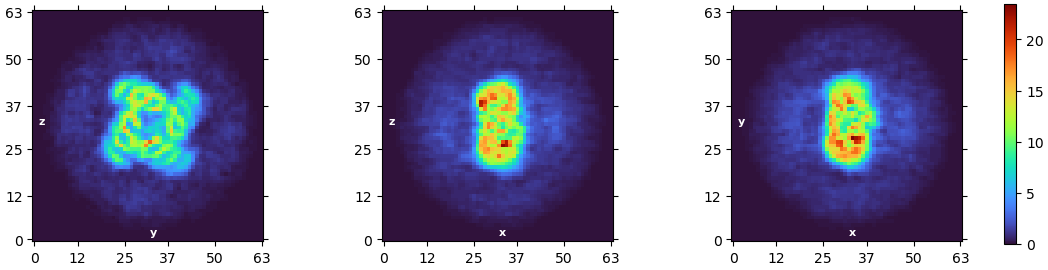
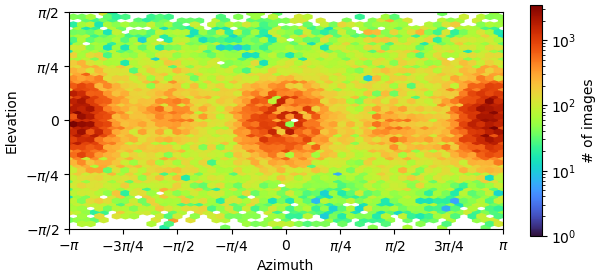
I’m currently facing a challenge in reconstructing my C4 homo-tetramer protein, which has a total weight of about 120kDa. Although I have obtained numerous top and bottom views, I’m encountering limitations when it comes to side-views:
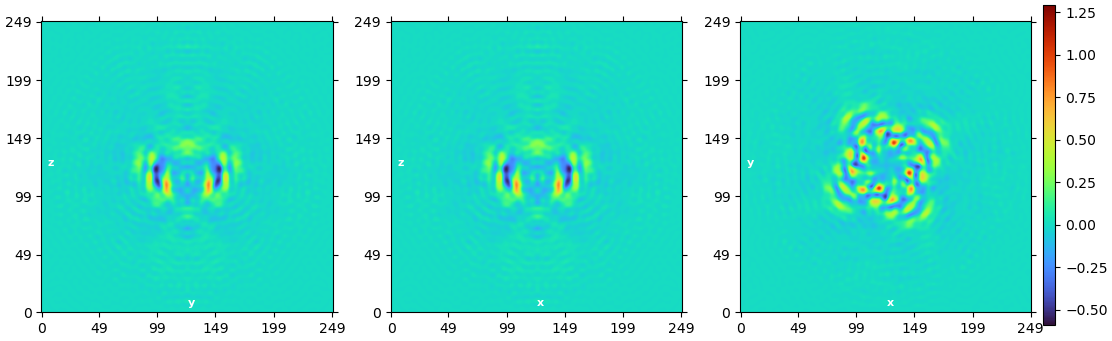
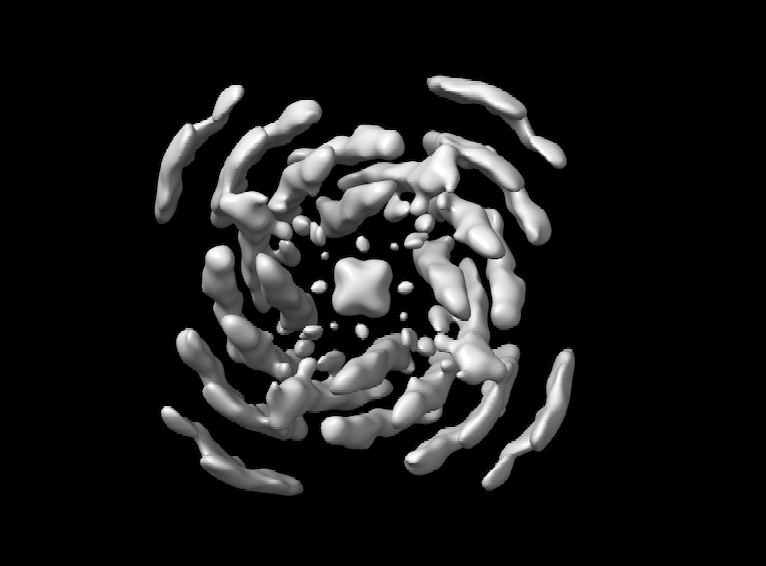
I have performed ab initio reconstruction and refinement, which resulted in a structure with a resolution of 5A (or 4.15A by applying symmetry). Unfortunately, the density of the reconstructed structure is of poor quality, even though I used 1.5M particles.
I suspect that this issue might be due to the preferred orientation of my sample, but I’m open to suggestions if I’m missing something crucial. So far, I have tried three different detergents and experimented with various grids. I was thinking of doing tilted collection next.
I would greatly appreciate any advice on how to approach this problem effectively.
Looking at your ab-initio, it looks like you have used only fairly low resolution information.
I would suggest trying with initial and final resolution of 12 and 9Å, respectively, as this is a fairly small oligomer and you have clear secondary structure evident in the 2D classes.
I wouldn’t be too worried about preferred orientation at this stage - in C4, you shouldn’t need all that many side views to get a reasonably isotropic map. Definitely worth training a Topaz model specific for the side views though, particularly if the micrographs are crowded.
When refining, you may also want to start at slightly higher resolution (15Å, rather than 30Å) as in my experience in cryosparc, for small proteins starting at too low resolution can result in incomplete convergence.
Also, looking at your map it looks like there are some mask-edge artefacts - I would try running NU-refine with dynamic masking switched off (you can do this by setting the resolution to start dynamic masking to 1Å).
I also followed your advice for refinement and the density does look better. However, I would say I am not there yet, regarding the helices near the edge
I also trained CrYolo picker with side views, which resulted in a substantial increase in the number of particles from 60k to 240k. I am currently re-running the reconstruction process using this new particle set
Glad it’s improving! I would consider the possibility that it may not be exactly C4 - consider the possibility of pseudosymmetry, with it being actually either C2 or C1.
To test this you could try a few strategies:
Refine in C4 → C4 symmetry expansion → 3D classification (no mask input)
Test refinement in C1 or C2, starting from the C1 ab initio.
Try ab initio with symmetry enforced (C2 or C4). It may take a couple of tries (different random seeds) to get the right symmetry axis orientation.
Alternatively, the poor density may be due to conformational heterogeneity (inter-subunit flexibility).
In that case, 3D-VA or 3D Classification may be helpful, starting from the C4-refined particle set, either with or without symmetry expansion.
Thanks for the advice, I tried some 3DVA and there might be some minor flexibility. Also performed C1, C2 refinement, but it did not help
An interesting observation I made is that when I perform NU-refinement, the edge density appears less compressed during the refinement process compared to the final density. It makes me wonder if there’s a way to address this using a mask.