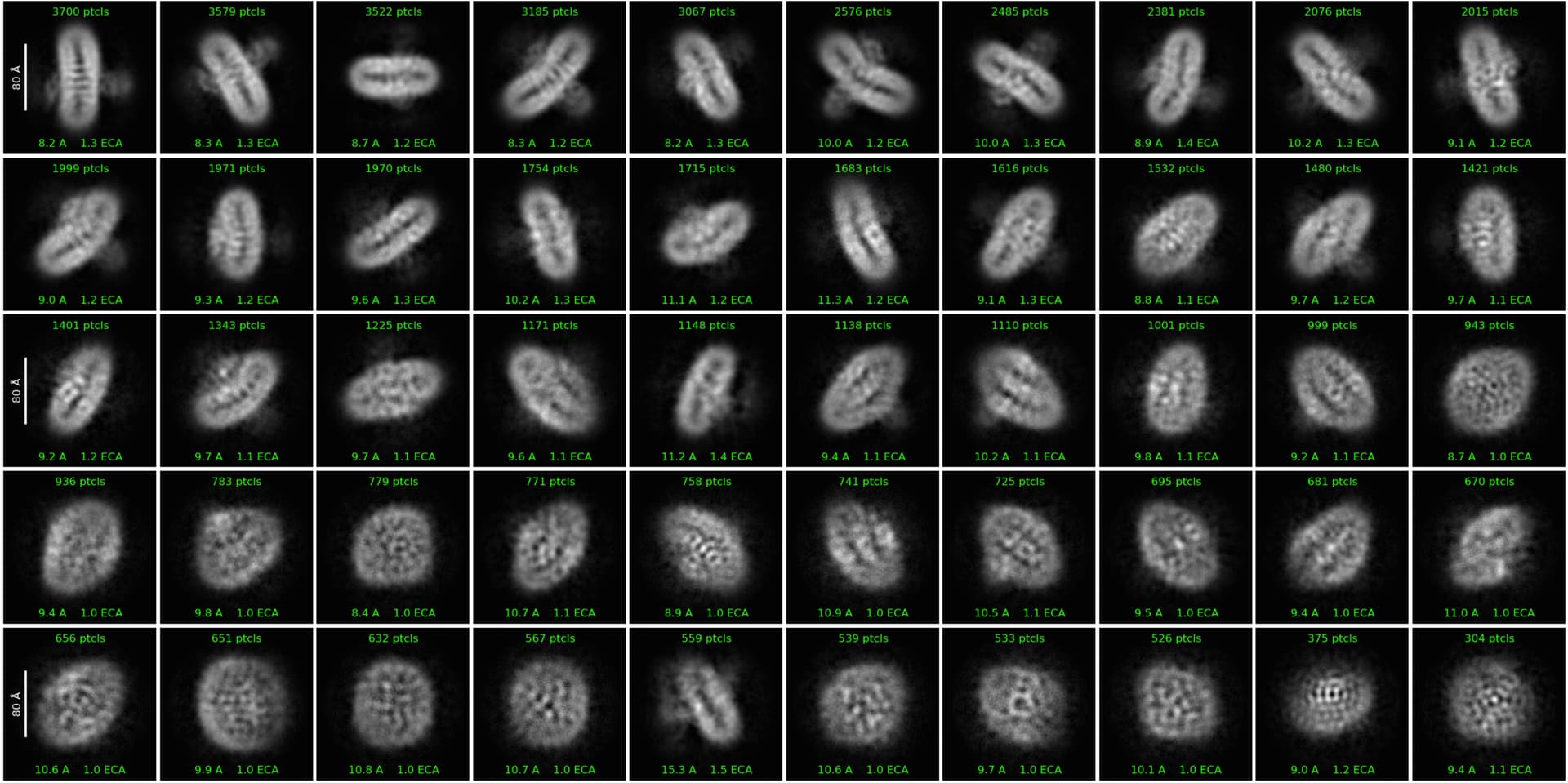
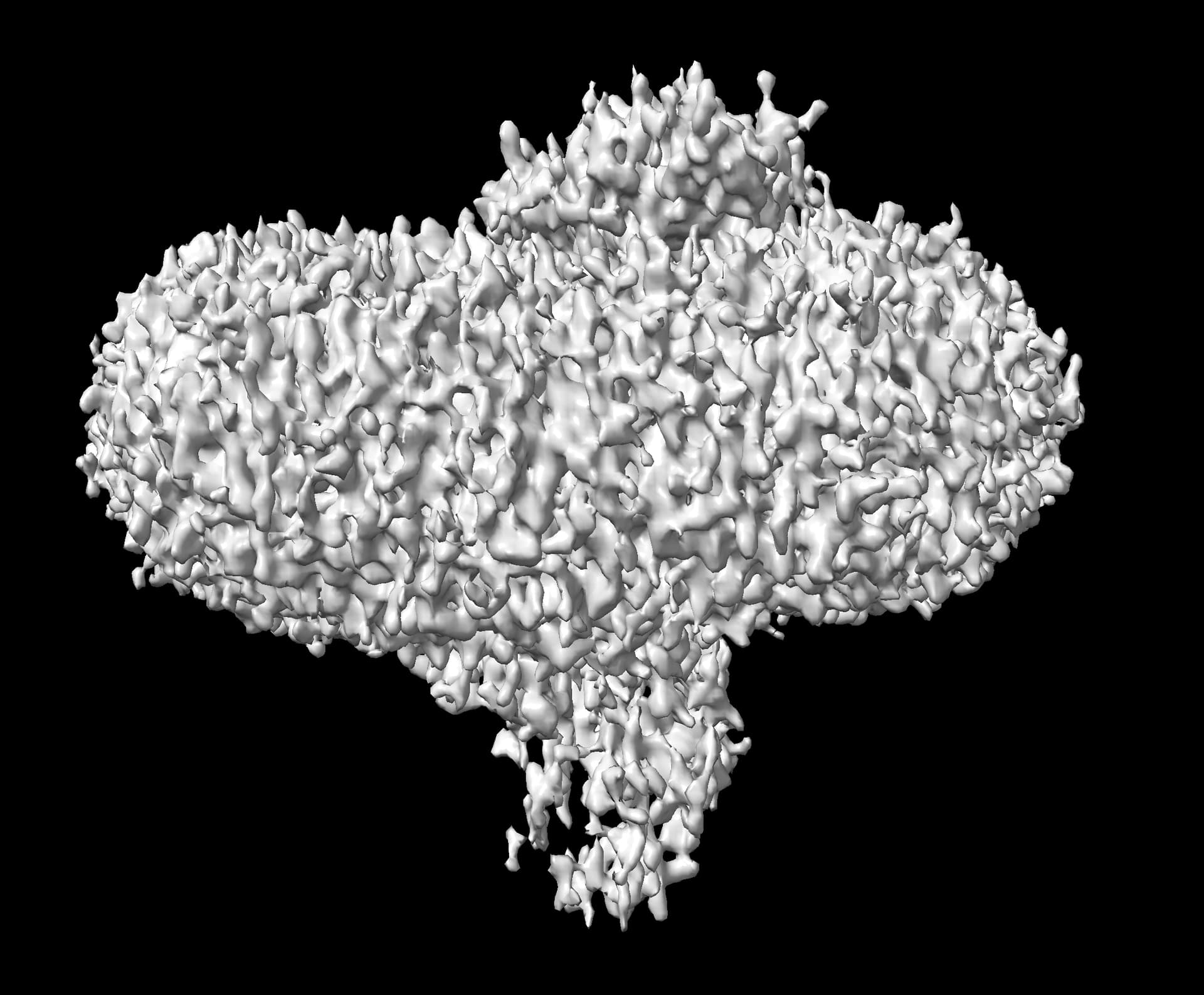
I am running an analysis on a membrane protein dataset and so far it has been quite challenging. I had to fine-tune a lot of things to actually get decent 2D classes:
The real problems though come at the next step. Homogeneous refinement, Heterogeneous refinement, Non-uniform refinement, all fail, usually resulting in maps of significantly worse resolution than the ab-initio model. I have never encountered a similar issue before, and I was wondering if anyone can offer some advice on the problem.
your box size is way too small. it should be 1.5-2x what it is now. You can re-extract with a bigger box and redo 2D classification. Try it with the default settings except these:
a mask that is the same diameter as your current box size
batch size 400
40-60 iterations
force max over poses/shifts: true and false
however many classes you want
You can try it with or without the non-negativity/clamp solvent.
How do your classes look with these settings and a larger box?
You shouldn’t set the final resolution in ab initio so high. It is not designed to get to 3A resolution, there are no half sets and you will end up just producing a map of noise (which is what your map looks like).
Also, how big is your protein? It also doesn’t look like you have many particles.
In addition to @Sia’s recommendations, I’d think about just using the first two rows of classes from the 2D you showed, doing another 2D and using only the very cleanest for starting Ab initio at 16-20 Ang and ending around 6-8 Ang. You can then use all of them for a heterogeneous refinement run into a moderate number of classes.