y sample contains two types of virus particles: one is the immature virus - empty capsid, which has no RNA inside, and the other is the infectious virus, which contains RNA and is infectious. I loaded the sample onto the grid and vitrified it using the Vitrobot, followed by imaging with a 300kV machine.
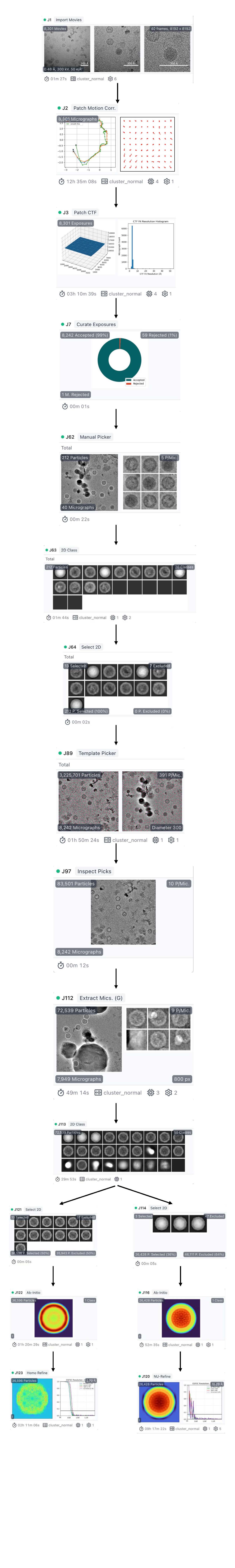
I imported the collected images into CryoSPARC, where I let CryoSPARC select two types of particles. Finally, I performed classification on the selected particles and separately picked the empty capsid and infectious virus to construct the 3D structures. The workflow is:
However, the final 3D structure of the empty capsid has a decent resolution (2.70 Å), but the resolution of the infectious virus particles remains low, regardless of whether I use homogeneous refinement or non-uniform refinement.
In this situation, how can I use CryoSPARC to improve the resolution of the infectious virus?
You should try CTF refinement (both per particle and global) for the empty capsid - looking at your FSC curves, and considering the particle size, you are probably leaving some resolution on the table. If resolution improves substantially (to nearer 2Å), you might consider Ewald sphere correction, also.
With regards to the empty capsid, does the ab initio model have the features you expect.? That would be my guess for where things are going wrong - perhaps heterogeneity of the contents are interfering with ab initio model generation. Have you tried using the empty capsid as an initial model for refinement of the full capsid particles, or playing around with the ab initio parameters?
Be careful when aligning to symmetry - sometimes during the initial phases of refinement, symmetry alignment can fail for highly symmetric particles, giving failures like you see.
Refining full things is fun. In my case, I was trying to refine a 260 kDa protein full of 500 iron atoms. I learned some lessons along the way. There are huge issues because of the domination of the diffuse density in the center of the particle that prevent good alignments ESPECIALLY when aligning to symmetry axes. I also wonder if the weak phase approximation starts to break down, although with RNA vs iron, I imagine that’s a bit less of an issue.
What I’d recommend trying is to take advantage of the icosahedral symmetry as much as you can. Mix in your full particles with your empty particles. Try to have 2 empty for every 1 full. Enforce icosahedral symmetry, and refine. If it goes well, you should see the resolution drop slightly compared to your empty capsid. But then you can run the particle intersect job, grab the particles that were full, and run a homogeneous reconstruction (not refinement) with symmetry enforced. Your resolution should improve.
Essentially, what this is doing is a roundabout way to downweight the contribution to the density from the RNA relative to the protein. You have innate 60 fold symmetry in the protein, but not the RNA, so you will get your best resolution if you can just align to those symmetry axes. Boosting that protein signal and downweighting the RNA should help.
Once you really good icosahedral alignments, you can now do a bunch of new tricks. You can subtract the RNA and try aligning the full capsids without that RNA present. This can help if you expect the full ones have minor conformational differences from the empty ones that you would not have found refining them together. You can also do symmetry expansion, to look for single subunit interactions with RNA. You can also do local refinements of the whole particle as C1, but not let one subunit move more than half the distance to the next nearest subunit, etc.
It’s not a perfect solution, but it’s the best one I’ve found.
Hi!
Your workflow is amazing!
Now, I am working with capside virus and has a icosahedral symmetry and is very big aprox. 740 A, so I want to know if I am on the good way. I have 48 000 particle and I did it homo-refine using default parameters and the result give me a 3.7 FSC, so Could you share me more ideas to continue?
Regards!