Hi all, I have a complex structure consisting of proteins A and B. I have got a high-resolution map of about 3.1 A. However, most resolution contributions from protein A. So I have tried to give a tight mask for protein B to improve the resolution of protein B. But it doesn’t work. Does anyone have other ways to improve the resolution of protein B?
Well I’d need a lot more information to try to help… starting from the biochemistry part, are you sure that B is always present? Can you see it in 2D classifications? What if you leave the high resolution aside for the moment, and try to see it in low res (say 7 - 18 angstroms max)? With 3DFlex you might try to check if the binding site is too flexible, but you’ll need a lot of particles.
My protein A is 200KD but B is 30KD. Actually, the intact particles(A bind B) only have 6% (48,000 particles). In the 2D classification, I can see B bind to A. I had run many rounds of 2D and hetero refine removed B un-present particles. only leave 30,000K particles(A bind B) and got a 3.1A map. And I know the binding site is very stable. Although I got a 3.1A map, the problem is protein A is 3A, and protein B is just about 5A.
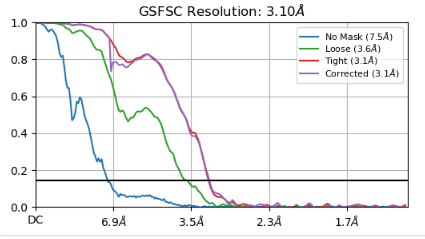
OK. Does the map really look like 3.1 angstroms (most side chains visible)? This is a bad situation for such a small set, you might consider going back to biochemistry and try to increase occupancy of B. If this is too harsh, you might want to spend more time rethinking the classification strategy, maybe focus more on 3D than on 2D classifications, trying to end up with more particles at the end. Then instead of trying to mask the little one, just inflate the mask on A until you start seeing B better. But I’m really betting more on biochemistry, and a larger set to begin with. - of course there might be people out there who think otherwise, so I’d like to read them, too.
If it’s a compositional problem, you’ll need to increase the % of A that have B bound. If it’s a computational problem, where maybe A is a lot bigger and thus dominating alignments, you should try particle subtraction, subtract A, and just refine B. But even then, B still needs to be big enough to refine independently.
Thanks for your suggestion. I’ll try more 3D classification. Maybe I need to scale up the data collection to ensure I have enough particles with Protein B.
Thanks for your suggestion. We will try the particle subtraction.
Hi @jasonhe!
In addition to the great suggestions above, I have two more things for you to consider:
First, you may want to consider using the Downsample Particles job to create smaller particle images, at least until you have your particle stack cleaned up. It looks like right now your Nyquist resolution is about 1 Å, which is much higher-frequency than you need right now. You could consider downsampling your particles by a factor of 1.5 or even 2.0, which would really speed up calculations.
For example, if your box size is 400px right now, I’d recommend trying downsampling to 300 or even 208 pixels.
My next suggestion is to try a 3D Classification job. First, use a Homogeneous Refinement to align all of your particles (ideally before they are classified with 2D) to a single map. This will likely result in a map with a high-quality A protein and a low-quality (or entirely missing) B protein.
Then, you can use 3D Classification to perform classification without alignment to separate out particles with and without protein B. 3D Classification accepts two masks: a solvent mask and a focus mask. I’d recommend trying a solvent mask around the entire protein (including any blurry region of protein B you can see) and a focus mask around the entire area where you expect to see protein B.
Since 3D Classification does not have to align the particles, it is much faster than jobs like heterogeneous refinement, and the mask lets you focus on just whether B is there or not.
I hope that helps!
2 Likes
Thank you @rwaldo.
That is a good suggestion for me. I will run the Downsample Particles job and then do 3D Classification. I will tell you the good news if it works for my data set.