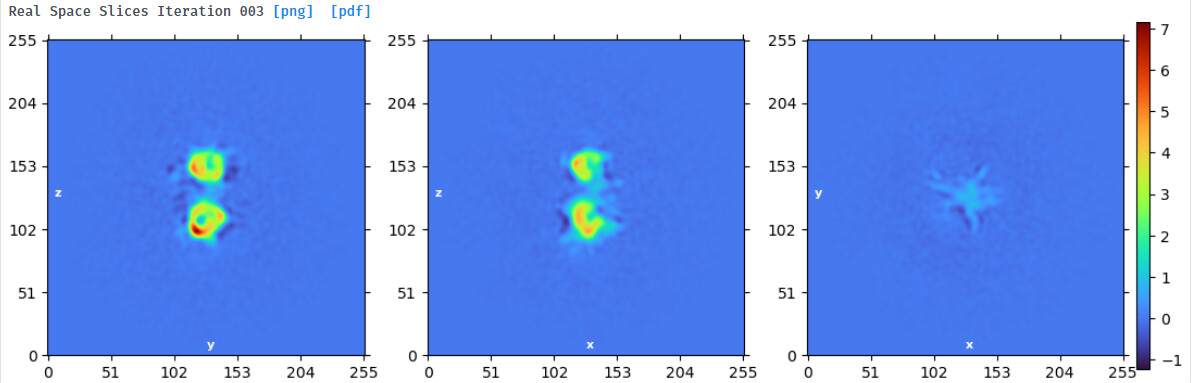
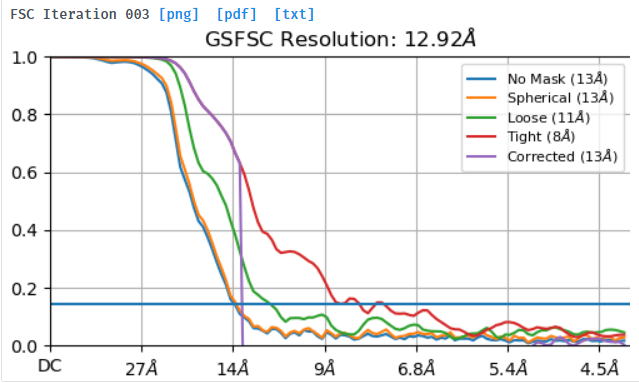
Hi,
I attended the Cryosparc v3.1 overview webinar this week and have a query related to a question that was discussed. I would really appreciate some help!!
When asked how to optimize the number of classes for ab initio 3D model generation, @apunjani recommended that you should test different values for K and you would know that it is too high when it separates different views instead of truly distinct conformations. During the webinar, Ali also commented that you should not aim to get the best 2D classes, but rather use 2D classification to sort particles from obvious junk and then take all protein particles into 3D.
After hearing this, I tried to run ab initio on all my 2D classes where only obvious junk was removed after Topaz picking.
This stack of 1.1million particles contains monomer and dimer - as seen by 2D classification.
The protein is difficult to work - it consists of 2 (monomer) or 4 (dimer) globular domains with a short linker, is rather small (140kDa monomer), flexible (within each domain and between domains) and N-glycosylated. The monomer is like a dumbbell and the dimer like a flat view of a butterfly.
Monomer side and top:

Dimer front:

Dimer top:

Dimer oblique:

Dimer bottom:

-
Previously, I only used 2D classes that looked like monomer to my eyes and ran that through ab initio. I got 3 different monomer 3D classes and 1 that looked like a single domain. Orientation distribution plots looked good for all 4.
-
When I selected all the 2D classes that I thought were dimer and performed 3D ab initio and refinement in Relion, I obtained a 4-domain dimer. But it suffers from severe orientation bias and there is likely flexibility in the non-interacting domains. Since 2D classification and human bias may have caused the missing views, I wanted to run the full post-Topaz stack through cryosparc ab initio.
-
When I now ran the full 1.1million particles through ab initio using different values for K, I consistently got 3D classes with only 1 or 2 domains. One of the 2-domain 3D classes is not a monomer but rather the two interacting domains of the dimer! I don’t see any 4-domain classes.
My questions are,
-
Can ab initio split the monomer into two separate globular domains so that it looks like a single-domain class? Or are there really single-domain classes in the data?
-
At low res, the 2-domain monomer may look like the top or side view of the 4-domain dimer (viewed from the ‘butterfly head’ or along the ‘butterfly wings’). Would the dimer therefore be pooled along with the monomer in ab initio? Would the particles redistribute between the classes as the resolution improves during heterogenous refinement?
-
Does 3D ab initio only show the interacting domains of the dimer because they are the most stable and thus easier to align? Or are the non-interacting domains (top ‘wings’ of the butterfly) missing because they are pooled with the monomer during ab initio?