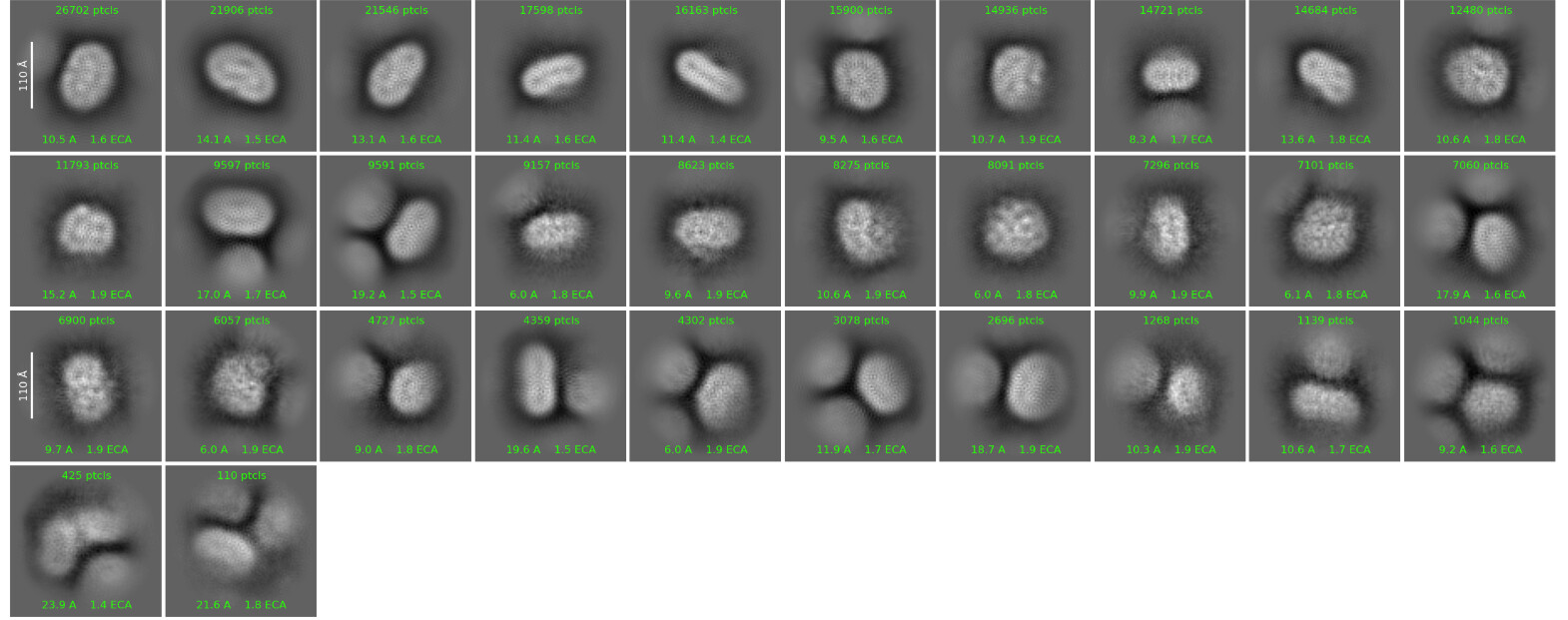
I am currently processing a membrane protein dataset. Visually, the particles appear promising in the micrographs. However, the 2D classification results are not ideal, and I am unable to clearly resolve secondary structural features.
Could this be due to misalignment? Does anyone have any suggestions?
Tighten the window up or impose a spherical mask so the neighbouring particles are not contributing to the alignments any more, do another 2D and increase interations, classes, target resolution and select classes which are nicely centred.
Then try ab initio into 4-12 classes, starting resolution 8, final resolution 4-6. I’m not usually a fan of doing this, but for small proteins which are entirely enveloped by micelle it is sometimes necessary as otherwise the micelle completely dominates alignments and you get junk.
Then heterogeneous refinement repeatedly until you either give up or get something reasonable.
Or go straight to ab initio from the extract job into 12-18 classes, starting at 12Ang or so and finishing at 6 or so.
Thank you for the suggestions, they seem very helpful.
I have tried applying a spherical mask, but unfortunately, the classification results remained largely the same as before.
Regarding the particle size, I wouldn’t consider my target protein to be particularly ‘small.’ The monomer is ~55 kDa, and I expect it to be a dimer (~110 kDa). However, does the difficulty in alignment suggest that the sample might have dissociated into monomers? Is it possible that the 55 kDa size is too small for accurate alignment with my current settings?
Additionally, if I am unable to obtain clear 2D class averages, does this imply that 3D classification is also unlikely to succeed? Is it worth proceeding to 3D steps?