I’m encountering an issue with 2D classification alignment in CryoSparc and I’d appreciate some guidance on how to resolve it.
I’ve collected data on cell vesicles containing multiple types of proteins and prepared single-particle cryo-EM samples. I’ve performed particle picking with oversampling to select the lipid bilayer of the vesicles and then conducted 2D classification in the hope of obtaining 2D information about a specific protein and reconstructing its 3D structure.
However, during the 2D classification process, the alignment seems to be based solely on the lipid bilayer, resulting in blurred average images of my protein particles. I’m seeking advice on how to overcome this issue and align the particles based on the protein instead of the lipid bilayer.
Could anyone provide recommendations on which parameters to adjust or any alternative approaches to improve the alignment and obtain clearer particle images?
The lipid bilayer is the strongest signal, which is why it’s aligning to that. You could try a very tight mask on the centre of the box as there are a few classes there which look like they might have some sort of extrinsic region of a protein aligned, or re-extract with a much, much smaller box size.
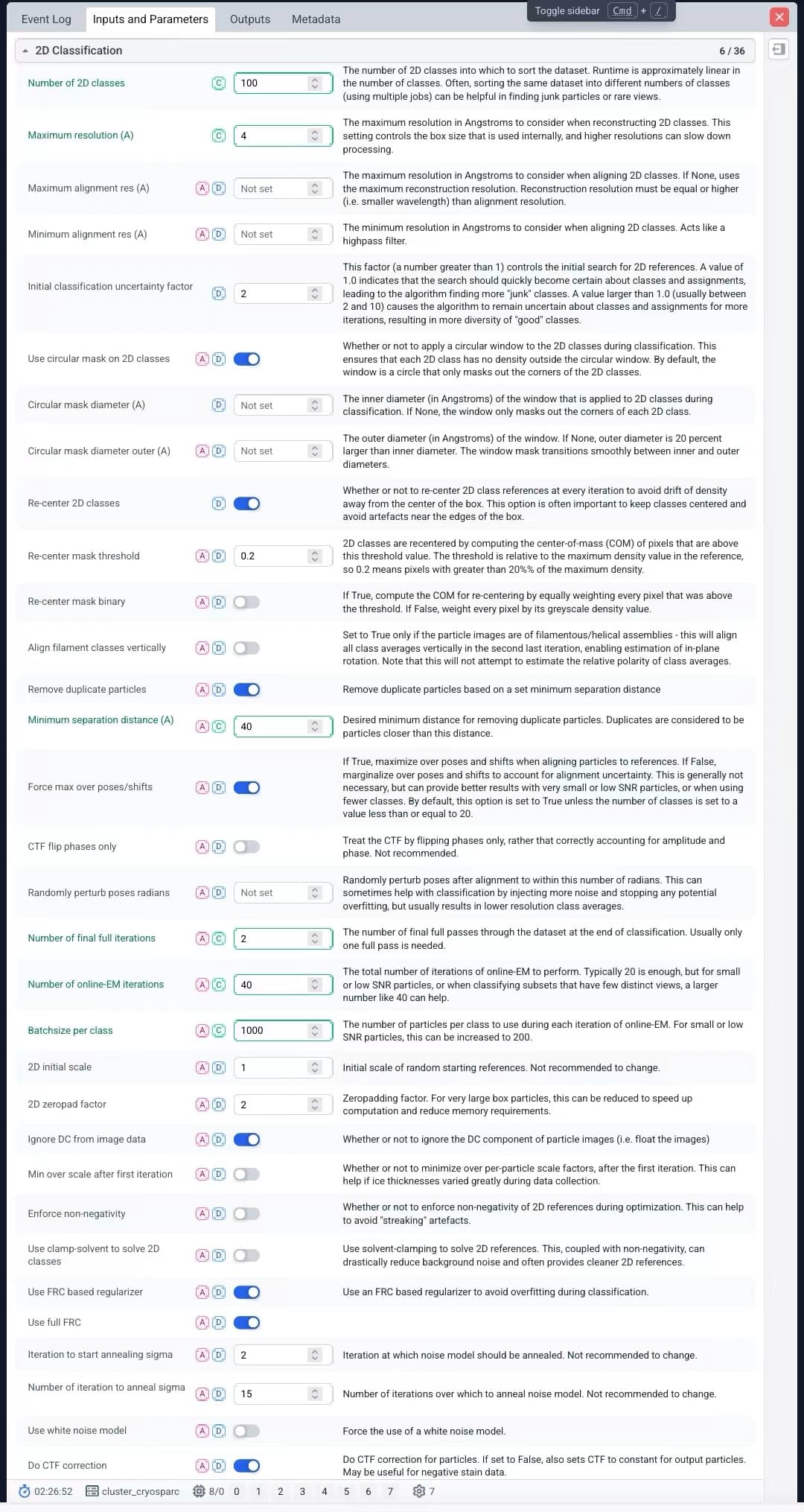
I’d initially try a much smaller box (maybe 20-25% the current size), 200-400 classes, classification uncertain increased to 5-8, 80-100 online-EM iterations and 3 full iterations. I’d back the Max res. back off to default, as I’m paranoid about overfitting when particle hunting (only increase Max res when cleaning an already reasonable particle selection). I’d also try turning off 2D class recentering. Also try turning off Max pose over shifts. And finally also try Enforcing non-negativity. Lots of things to try, will take a few runs to work through them all.
Without details on size/mol weight of your target, acquisition angpix and box size, it’s a little difficult to say further.