Sample Description: I have purified vesicles from cells and aim to resolve the structures of membrane proteins embedded in these small vesicles.
Issue: I initially picked particles located on the vesicle membranes using an “oversampling” method and proceeded with several rounds of 2D classification. However, each classification result aligns with the vesicle membrane instead of the membrane protein, leading to blurry protein images in the 2D classes.
Attempts to Solve:
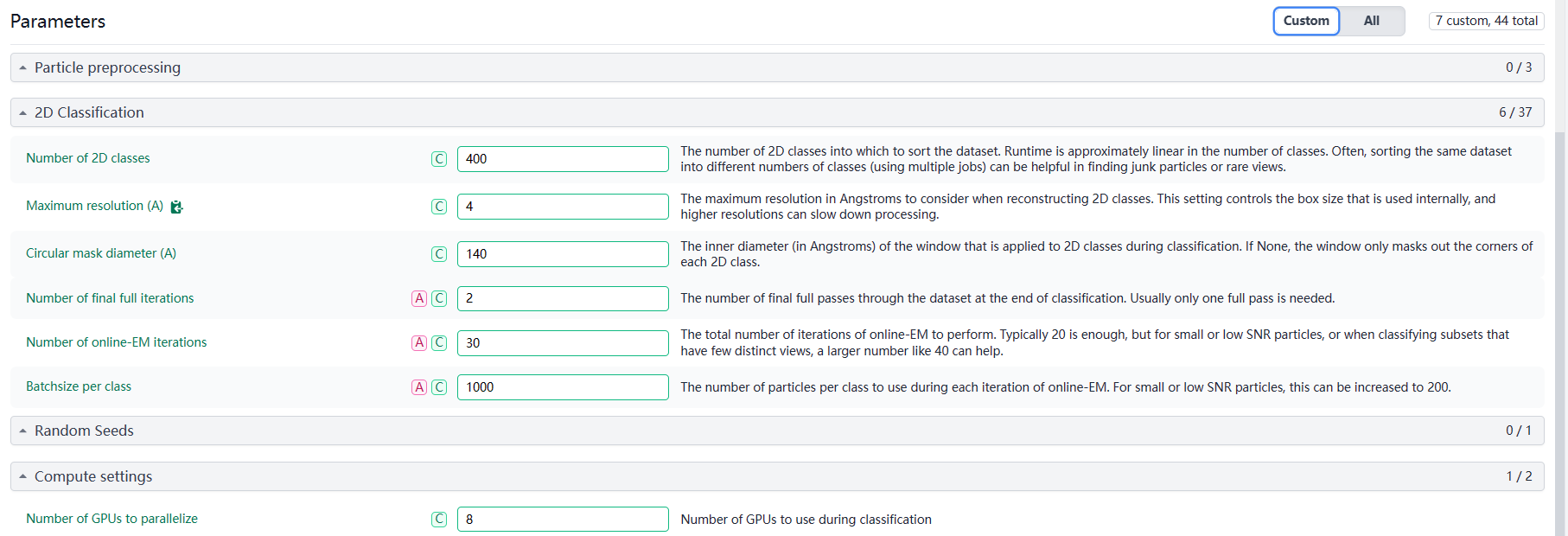
Extraction Parameters: I used an extract box size of 180 pixels and a pixel size of 1 Å.
2D Classification: Despite multiple rounds of 2D classification, the alignment focuses on the vesicle membrane rather than the membrane proteins.
Try a much tighter mask. Also try a multi-class ab initio reconstruction followed by multiple rounds of heterogeneous refinement. Usually the micelles are fluid enough that they cancel in 3D and the protein can start to dominate.
I will try using a much tighter mask to focus on the membrane proteins.
Regarding the 3D reconstruction, I often encounter an issue where the alignment focuses on the vesicle membrane, resulting in an artifact where it looks like a vertical rod is inserted into the membrane.
Do you have any additional suggestions for addressing this issue during the 3D reconstruction process?
I guess it confirms at least that you have the correct alignment, and you just need to sort more particles. Or a not too generous particle subtraction might help. I am just guessing here.
In fact, we have been trying to find particles that align correctly. A while ago, we successfully resolved the structure of one of the larger proteins on these vesicles, which was relatively straightforward due to its size. However, we still have some smaller proteins that need to be resolved. After several rounds of 2D classification, I still cannot see a sufficiently clear class of aligned particles.
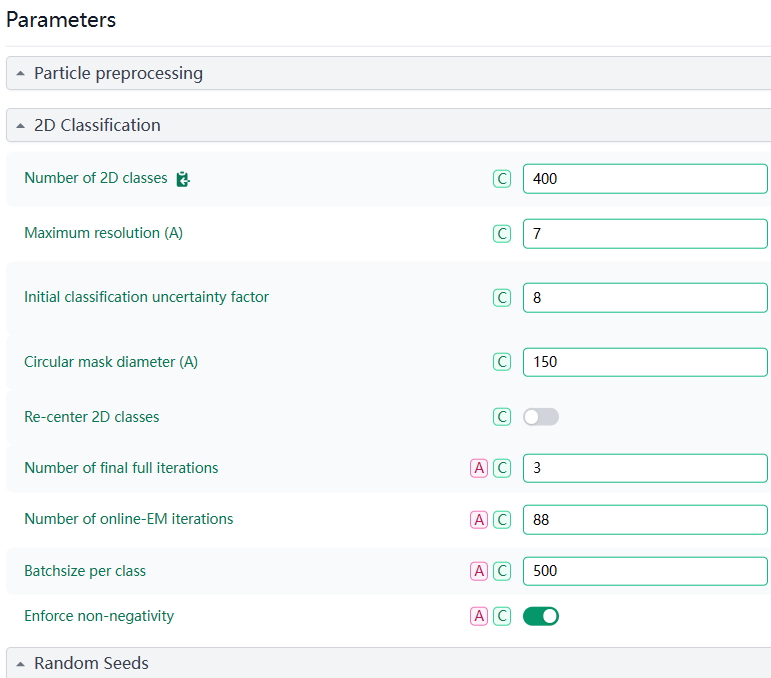
My basic approach is that once I see a relatively clear 2D class, I would isolate it for further processing. However, so far, I keep getting unclear 2D classes. Here are the results and parameters from my latest round of classification.
How many particles do you have in your best ab-initio and overall?
It is difficult for me to say anything about your parameters, they look fine. I usually don’t value full iterations, they do nothing that I expect from 2D classes but slow down the processing tremendously.
I worked on small membrane proteins (ca 50 kDa structured part), and in the end I managed to solve the structure without a fiducial marker. Nothing new nowadays, but I was never able to obtain great 2D classes. I just used it to sort out obvious junk, and went on for further 3D classification using exhausting ab-inito jobs and heterogenous refinements.
I haven’t tried this on such a case, but perhaps you might try using a 15 or 20 Å highpass filter during 2D? I am assuming the contribution of the membrane should be greatest at low res, and less at higher res, whereas hopefully your membrane protein will have more signal in the secondary structure range (12-6Å or thereabouts. I have tried this on other systems with decent results - it does take a long time to converge though, so if the classes look like noise for quite a few iterations just give it some time.
Thank you for your response and sharing your experience.
Regarding your question, in my last 2d classification, I have more than 1,000,000 particles, and overall, I have not tried ab-initio reconstruction.
It’s reassuring to hear that the parameters seem fine. I appreciate your point about full iterations; I’ll consider reducing them to speed up the process.
Your experience with small membrane proteins is very helpful. It’s encouraging to know that achieving great 2D classes isn’t always necessary and that sorting out obvious junk before moving on to extensive ab-initio and heterogeneous refinements can still lead to success. I will follow this approach and see if it improves my results.
I will try using the highpass filter in my next steps. I have also read Jack Zhang’s paper and listened to his recent online talk. Additionally, I came across another paper published in Science titled “Molecular mechanism of the ischemia-induced regulatory switch in mammalian complex I.” I plan to try the methods discussed in these papers as well.
The vesicle membrane subtraction can be done using Fred Sigworth’s matlab scripts:
(K. H. Jensen, F.J. Sigworth and S. S. Brandt, “Removal of Vesicle Structures from Transmission Electron Microscope Images”, IEEE Transactions on Image Processing, 2015; 25(2):540-552.
K. H. Jensen, S. S. Brandt, H. Shigematsu and F.J. Sigworth, “Statistical modeling and removal of lipid membrane projections for cryo-EM structure determination of reconstituted membrane proteins”, Journal of structural biology, 2016; 194(1):49-60)
Overall, small protein with membrane datasets are challenging. If the protein itself is too small to be anlyzed by cryo-EM, with membrane it just makes things more difficult. For large proteins, depends on the target, the membrane subraction maynot be necessary.
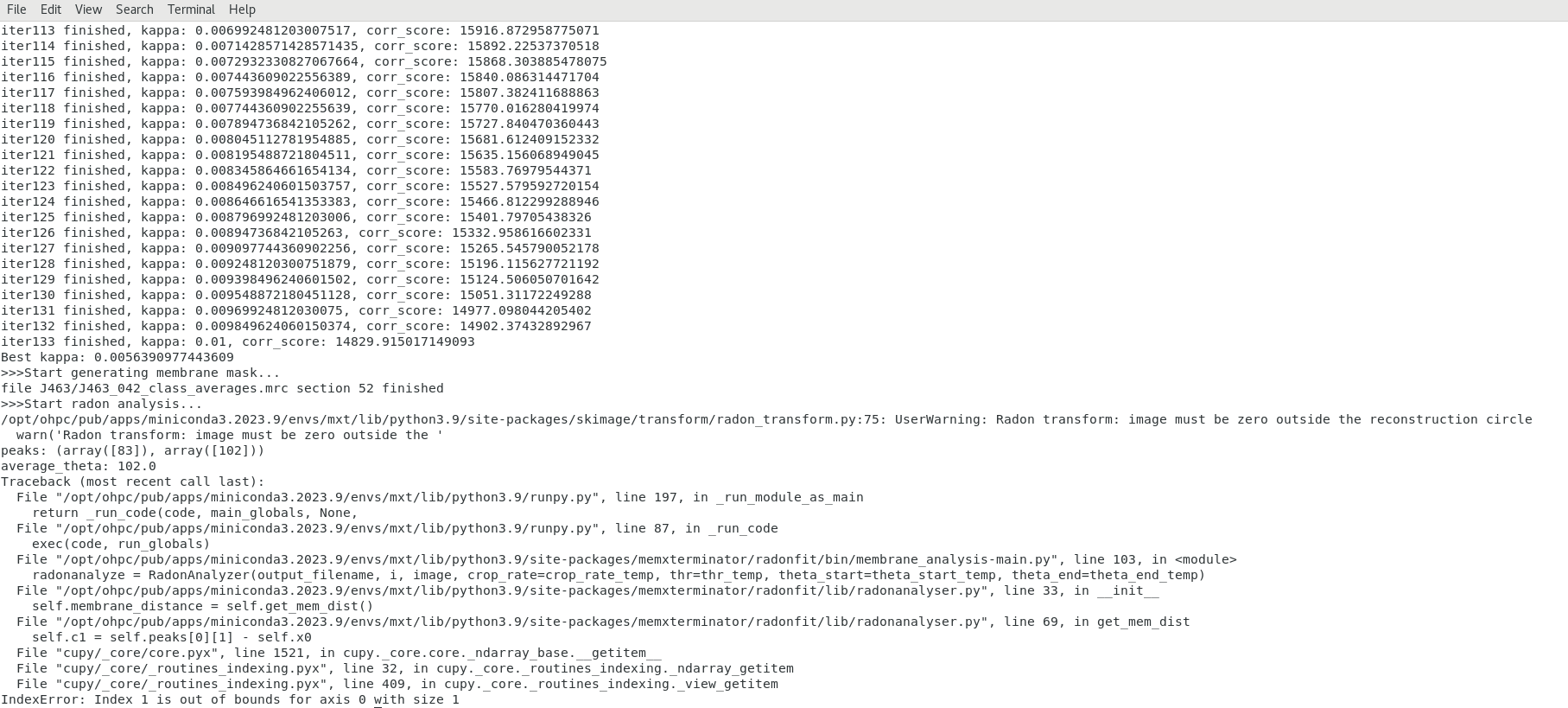
I noticed the Cryo-MEM link you shared in the Zoom meeting chat last time and gave it a try. However, I encountered an error during the second step (radonfit). Please see the attached screenshot for the error message. I will try the other two methods you mentioned as well.
Looking forward to the updated version of Cryo-MEM.
Also, you membrane 2D class averages look very homogenous. You can also try to reconstruct the 3D volume of membrane (treat these membrane as your targets) and then subtract them using standard post-3D subtraction methods.
I appreciate your advice on the homogeneous 2D class averages. I am particularly curious about the rods I see angled into the membrane; I am not sure what they are. Our candidate protein likely has a tertiary structure composed mostly of α-helices. However, I think it is worth trying the 3D reconstruction. I will proceed with ab-initio reconstruction and heterogeneous refinement in CryoSparc.