Hey everyone!
I am currently trying out masking for the first time, using a dataset of a relatively small protein with a binder attached.
Since I have an inkling where the binder might be from 2D classification, I’ve created a mask for that region as well as one excluding it using the volume eraser method outlined in the tutorial.
Unfortunately, attempting to run a local refinement using the mask of the region of interest keeps failing during the initial model creation:
Traceback (most recent call last):
File “cryosparc_master/cryosparc_compute/run.py”, line 129, in cryosparc_master.cryosparc_compute.run.main
File “cryosparc_master/cryosparc_compute/jobs/local_refine/newrun.py”, line 430, in cryosparc_master.cryosparc_compute.jobs.local_refine.newrun.run_local_refine
File “/home/lab/opt/cryosparc/cryosparc_worker/cryosparc_compute/plotutil.py”, line 310, in plot_central_slices
slice_yx = newfourier.convert_herm_to_full(M[(N//2),:,:]) if herm_volume else M[center[2],:,:]
IndexError: index 259 is out of bounds for axis 0 with size 256
But running the same job with the same parameters using the complementary mask works. Could anyone give me some pointers on how I might be able to fix this?
Hi @LJS! Welcome to the forum! Could you please let me know the following information?
How did you make the mask, after using the volume eraser tool in ChimeraX? For example, did you upload the volume, import it with Import 3D Volumes, and then add a soft edge with Volume Tools?
What is the size of the mask, in voxels? If you download and open the mask used for Local Refinement, this should be visible in the log (see below for an example). We’re looking for the grid size numbers, but the pixel size might be relevant too.
Hey @rposert, thanks for the welcome! And my apologies for the late response.
You’re already on track with the first point, I did somehow forget about adding the soft edge. I guess I got lucky one of the maps still worked?
I am trying to figure out how to correctly add the soft edge at the moment, but im focusing on a rather small part of an already small protein - do you by any chance have any pointers on how to best set the thresholds for such cases?
Hi @LJS ! It is important to add a soft edge, so I’m glad we caught that, but I’d be surprised if a missing soft edge is what’s causing the issue here. I’m more curious about my two questions above – what was the exact process you followed to create the mask (what jobs, what parameters, etc) and what is the box size of the mask?
As for guidance on thresholds and the like, it will depend on the size of the domain and the target. We have some general thoughts and guidance in the guide, especially the Local Refinement page and the yeast spliceosome case study which might be helpful!
To answer your second question, this is the log when downloading and opening the mask in ChimeraX again:
As to the process, I’ve tried to follow the mask creation tutorial as much as possible.
I attempted using volume segmentation, but for some reason the selected region would always remain empty, so I moved on to the method using the volume eraser.
I did this exactly as described in the tutorial, creating first the mask for my region of interest, then the subtraction mask.
Saved both as an .mrc, scp’d them to the workstation and then used the Import 3D Volumes job with ‘mask’ as the type of volume to be imported.
I then tried using both masks for local refinements(without adding the soft edge), which worked for the substraction map, increasing the resolution by 0.4 Å, but repeatedly ran into above-mentioned error when using the mask for my actual target region.
Now that I am trying to figure out the soft edge part, the refinement is working but gives a warning (‘WARNING: Detected NaN/INF in 256 / 256 particles or frames:…’) and the resolution jumps to some 160 Å.
Not sure if this plays a role, looking over the mask again I saw some dust-like parts, I will give it another run removing those, but since I’m wondering whether my binder is here somewhere, I was hoping to avoid this.
Hi @LJS, good to know your mask is the right size. For the next steps in trying to figure out what’s going on, it would be helpful if I could see the screenshots from the Volume Tools jobs you used to make your masks, or images of the masks themselves. Also, approximately how large (in kDa) is the region inside your mask?
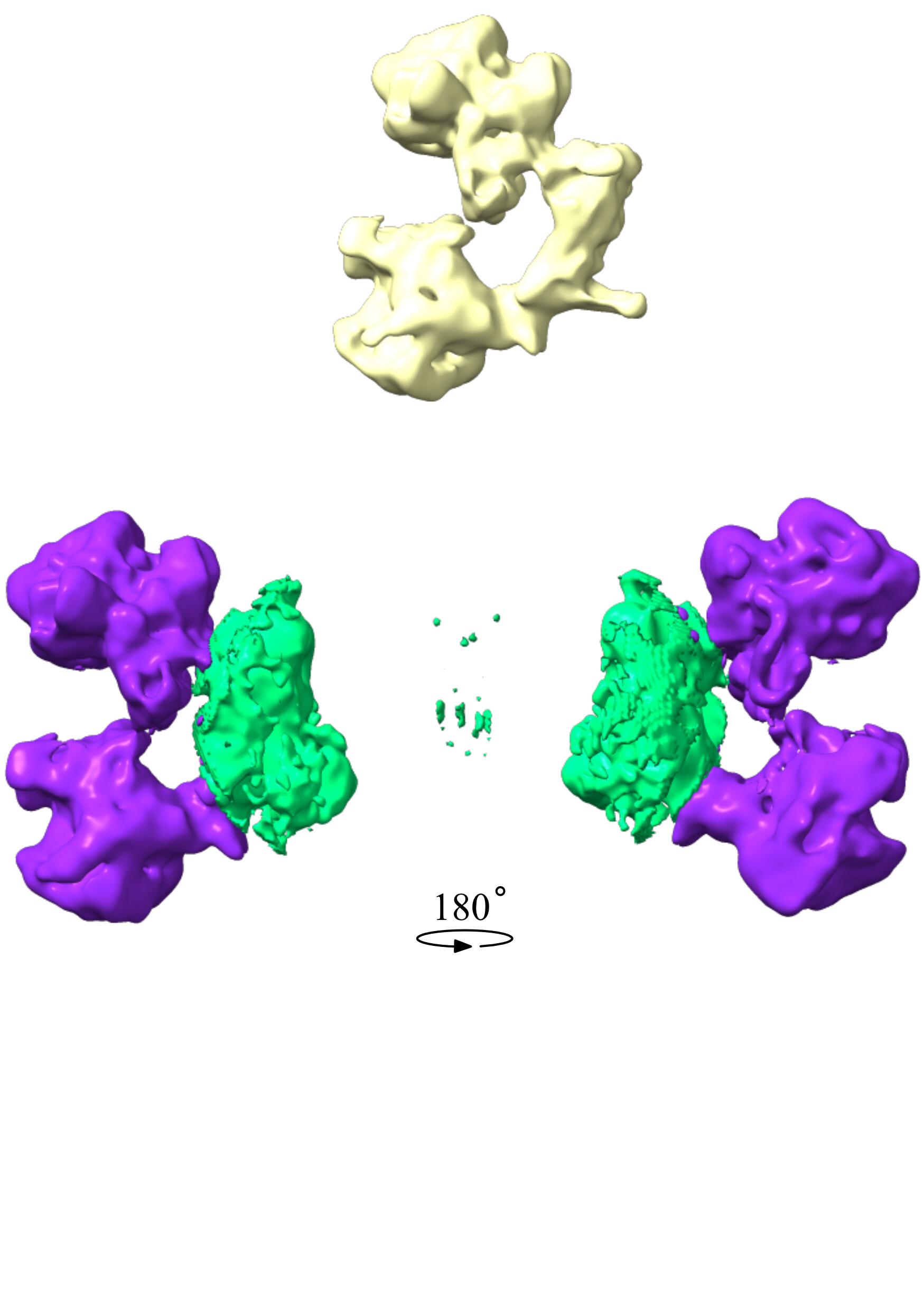
Here’s the blurred volume I used to generate both masks, as well as a look at either of them. The one coloured in green is the one giving me trouble, the purple one is the one that worked.
One thing I’ve also noticed know while looking at them is that erasing things outside the sphere changes the default contouring of the mask. Might be my ignorance talking, but does that have an impact on the refinement? The same doesn’t happen to the other mask after all.
Plus, this also happens when redoing the whole process.
The region I am isolating via the mask is around 12 kDa, out of the total 70 kDa that the particle should be - without the binder that I am looking for.
It’s not immediately obvious to me why these masks are causing the error you reported above. However, I do want to make a few observations – maybe when we fix these things that error will resolve.
First, I just want to say that 12 kDa is a very small subdomain to mask out. I am not saying it won’t work, but it will be very challenging! If you’re using a mask and Local Refinement to model movement, you may have more success with techniques like 3D Variability Analysis.
As for your masks themselves: your purple mask looks pretty good to me – it’s smooth and doesn’t have too may high resolution features. Your green mask, on the other hand, has two issues I can see.
It has extra “dust” floating outside the main body mask. I would be sure to erase this before using this to make a mask, as these unconnected regions can cause problems.
It has a lot of “bumpy” and “spiky” features, perhaps from noise in the underlying map. You may find more success by, after you erase the dust, using volume gaussian #{id} sd 2 (where id is the number of your volume in ChimeraX) to blur out these features. Often Gaussian filtering makes the volume slightly smaller, so you may need to increase the threshold contour or add more pixels of padding when producing a mask.
If you like, why don’t you give those a try and see if they help with your original problem. I’m happy to explain more if any of this is unclear, or if it’s not clear why it might help. Designing masks is a critical but challenging skill, so I’m happy to help you figure your way through it!
The main reason im using such a small part of the protein is because I thought I saw some densities in this region during 2D classification which then disappeared, so I wanted to investigate whether my binder might be ‘averaged out’ during the later refinement processes, and I didn’t know whether the variability analysis would work that way.
Thanks for the suggestions on improving the masks, the dust was something I also wondered about due to the tutorial - I’ve given both your suggestions a try now, subsequently, but sadly the error still appears (without adding a soft edge so far).
One thing I found a bit confusing is why the contouring is so drastically different for the one mask, showing all those spikey features as you also mentioned, while the other one does not.
Based on the answer you’ve given on the forum previously (Question about how masks are used - #2 by rposert), I understand that’s not something that can be changed by adjusting the thresholds in ChimeraX, but is actually done by applying the soft edge correctly, right?
So I guess the last thing to try is to get that right somehow. Is there any faster way to check the resulting masks other than downloading all attempts and opening them in Chimera again?
In any case, thanks a lot for taking a look at this!
Yes, if you add padding and a soft edge higher-resolution features (like the spikes) will tend to disappear. If that’s what you mean by “different contouring”, then yes, adding padding and a soft edge will help! However, I generally prefer to get rid of high-frequency features by first lowpass filtering the map, then creating a mask from the lowpassed map.
I (and others on the team) have actually found 3DVA can perform quite well in cases like this! It’s always worth a try! You could even just make a spherical mask around your entire protein (so that you don’t have to worry about the dust and spikes) and see what a 3DVA job with two components and a low filter resolution (maybe 14 Å) finds. It might not work, but it might find the features you’re looking for!
Could you please copy and paste the new error here like you did above, and also confirm for me that both your mask and your map are 256 x 256 x 256 pixels? This is a surprising error to me, so I’d love to get to the bottom of it!
I’ll keep playing around with the volume tools, so far finding the right threshold is proving rather tricky.
Doing a 3DVA this way sounds interesting, I’ve run a previous one and couldn’t find any new features, but that Job didn’t have the same set-up either - I’ll give that a go too, thank you!
The reappearing error seems to be basically always the same, just with complains about different indexes.
Both mask and map should have the right size based on the screenshot above, yet the following error appeared for all new masks I’ve tried:
Traceback (most recent call last): File “cryosparc_master/cryosparc_compute/run.py”, line 129, in cryosparc_master.cryosparc_compute.run.main File “cryosparc_master/cryosparc_compute/jobs/local_refine/newrun.py”, line 430, in cryosparc_master.cryosparc_compute.jobs.local_refine.newrun.run_local_refine File “/home/lab/opt/cryosparc/cryosparc_worker/cryosparc_compute/plotutil.py”, line 310, in plot_central_slices slice_yx = newfourier.convert_herm_to_full(M[(N//2),:,:]) if herm_volume else M[center[2],:,:] IndexError: index 323 is out of bounds for axis 0 with size 256
Let me know if there’s any other information that could help!
Thanks for including this job report. I was able to look at it alright, although this appears to be the job report of a 3DVA job that is failing for a different error. If you have access to the original local refinement that failed due to the IndexError, are you able to upload this one instead?
My apologies, that was indeed something else!
While I don’t have the original job report since I’ve tried to rerun the job with different inputs, the error has been easy to reproduce, so if that’s alright all provide the latest attempt with the same index errors. Hopefully the right file this time. J89_Local_Refinement_job_report
It appears you are using an input mask directly from an “Import 3D Volumes” job. Could you try the following:
Download the volume output by the Import 3D Volumes job
Open the volume in UCSF ChimeraX
Adjust the threshold until you are satisfied with the contour level; note the threshold value
Create a Volume Tools job
Connect the imported mask to the input mask slot
Set “type of input volume” parameter to mask
Set “type of output volume” to mask
Set “threshold” to the noted value
Set “Soft padding width (pix)” to 12
If the job completes successfully, can you let me know if you are able to download the output mask and open it in Chimera, and it still reports a cubic shape?
If so, can you re-try Local refinement and let us know if the error still persists?
Sorry for the long wait.
I’ve finally had the time to go over this, and it does fix both the Volume Tools as well as the Local refinement issue - thanks a lot!
I’ve also discussed things with some local experts, and realised the whole mask-creation works much smoother when setting the Import Volumes Job to import a map instead of a mask. I thought I was importing a mask already, not that the Volume Tools would turn it into one. I guess there was a bit too much confusion on how to make this work just based on tutorials.
In any case, thanks to both of you for looking into this!