Hi all,
I am using cryosparc 3.2.0 to get the structure of a protein complex (composed of two different proteins) and has difficulties to make sense of the map I got, although the resolution looks quite promising.
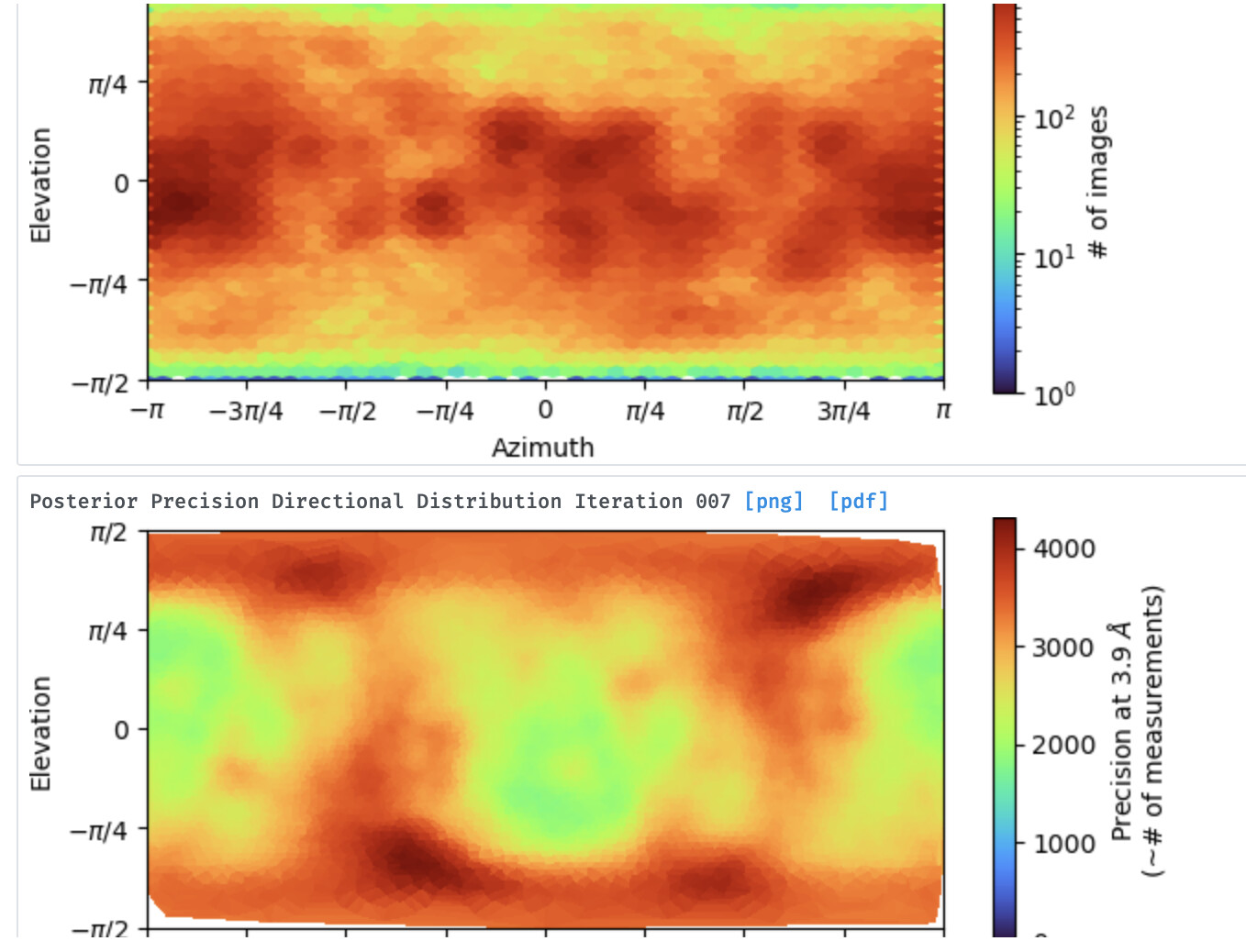
I initially selected around 875,000 particles from 2D classification, then an ab initio run with 2 classes, following a hetero refinement and I then run refinement with the best looking class (around 640,000 particles) which gave a map around 3.0A (applying C1 symmetry). However, the map looks a bit odd from one side (It has a sort of bump). Considering the class of this protein family, I expect a symmetric solution. Indeed, when I look at some of the images from 2D classification, I could really count obvious corners. I also tried to impose symmetry (such as C3, C6, D6) during hetero refinement and refinement steps, yet it resulted in worse resolution (>5.5A). In addition, I run ab initio job using the best class above (640k) and tried to divide into more classes (3 different) and run hetero refinement, but the solutions I got is more or less the same. Finally, I also picked less amount of particles from 2D classification which looked quite clear, but again one side the complex looks a bit unexpected to me
.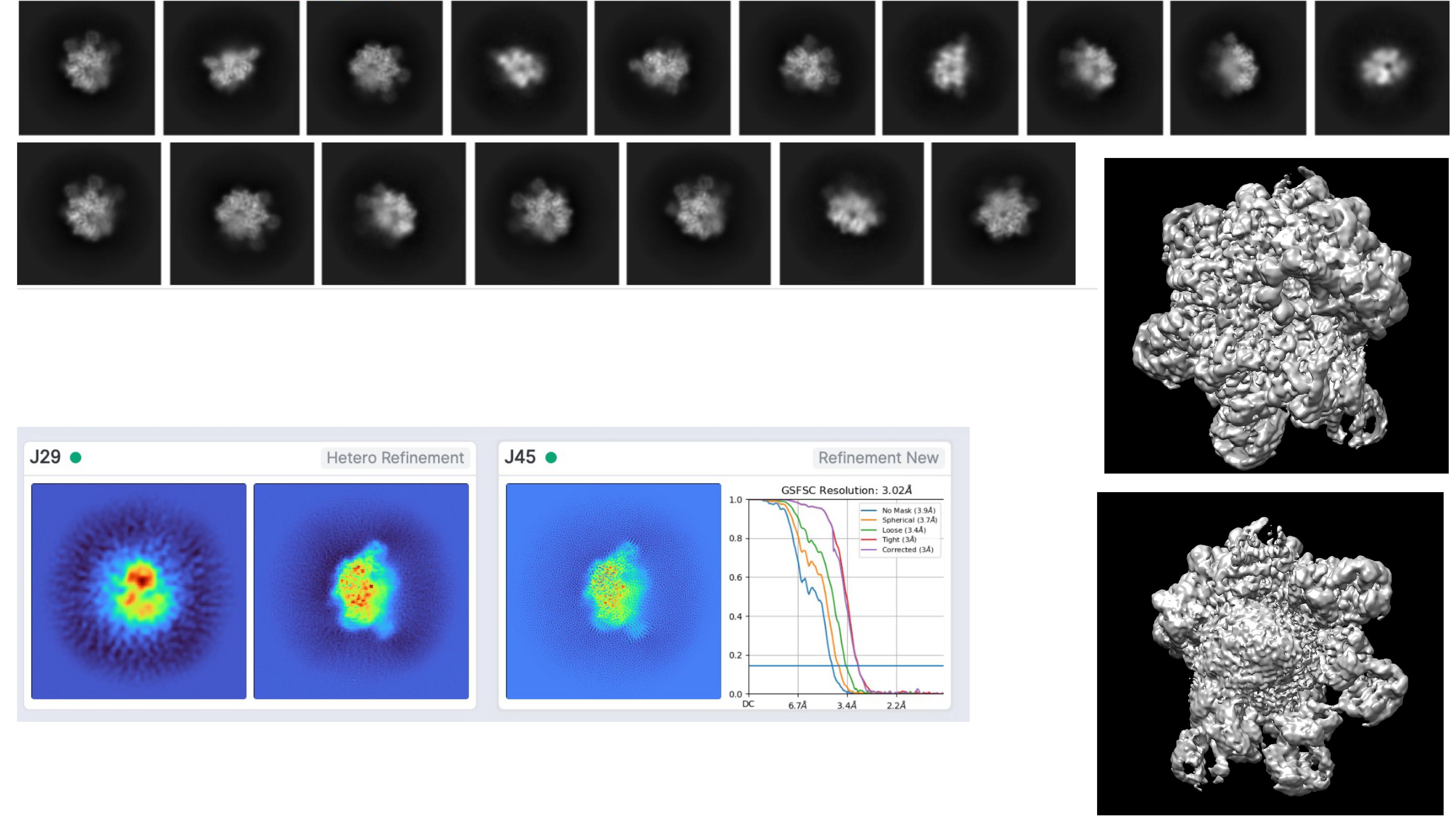
I am relatively new to cryo-em. So, I will highly appreciate if anyone could give any insights what might be wrong or what I am missing. Perhaps,
-
Not enough views (info) for this side of the complex, so couldn’t construct that part?
-
Symmetry problem? (I highly would doubt, I tried to impose all the available symmetry options)
-
Has to tune ab inito reconstruction parameters? (I run several runs by changing initial and final minibatch size (increasing)) and initial learning rate.)
-
Perhaps, it is what it is!
Thank you very much!
Best,
Esra