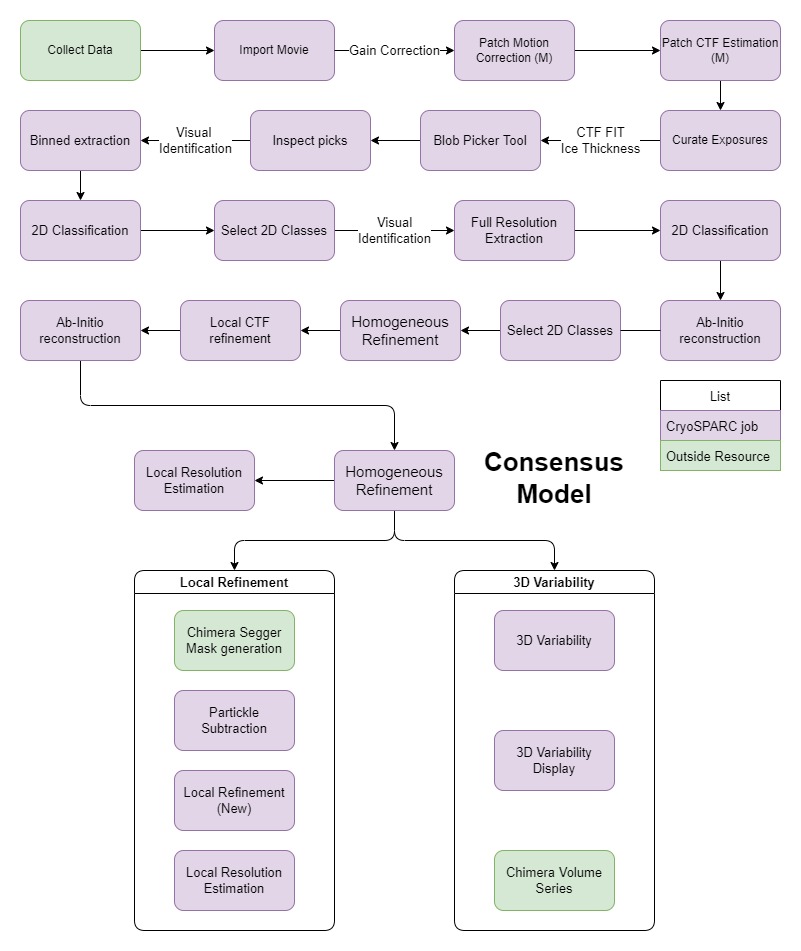
Hi all!
I’m an undergraduate student new to the subject. I’m currently working on implementing cryo-EM analysis capabilities in our lab using CryoSPARC. I’m also new to the subject as a whole. Using the fantastic online course by prof. Grant Jensen and the documentation provided by CryoSPARC I built an analysis protocol that looks like this:
Using this protocol, we achieved a resolution of 2.9A, which is pretty good for a first-time try. However, when I Attemped to incorporate a global CTF refinement step or a local motion correction step, the resolution didn’t improve or even degraded. I am also not sure if I used every job in the correct order.
From a quick look at this forum, it is clear that I have a lot to learn on the subject. Any input you may provide will be very appreciated.
There’s a thread somewhere around here about whether or not local CTF refinement will work. From my understanding, Relion seems to be the program of choice for things like local CTF refinement and particle polishing, although it’s a pain to try to get the particles imported and I haven’t had experience with it.
Your workflow looks very clean- mine would likely look like spaghetti when I first start with a new molecule. There’s a lot of going back and forth trying to figure out which parameters my dataset is sensitive to, and in my case I’ve had difficulties dealing with a very flexible molecule that has difficulty centering for something like a blob-picker. So there are lots of tricks out there. Sometimes I do multiple ab initio models and take each model and do 2D classification on the particles to get out any sneaky noise. Heterogenous refinement has made a huge difference for me with all of my flexibility.
I think it would make more sense to do 3DVA before downstream refinement steps if you are planning on pulling out multiple conformations from the 3DVA. But it would be interesting to see what others have to say. I’ve only been at this for a couple of years.
I was lucky enough to get a ribosome as the first structure to work with, so most of the defaults seem to work. Hopefully, I’ll be able to use RELION soon for the particle polishing and CTF corrections there. And I should probably fork 3DVA upstream from where it is currently. The server did not enjoy getting the full box size, and I had to add a downsample job.
Same here. SO much faster with much less crashing. You can look at the Fourier radius v box size to get an idea of how much you can downsample without losing resolution. There’s another post somewhere about that. As long as the radius is no more than 80% of the Fourier box size you’re good, or something like that.
@lutzkyguy Your scheme is very good! Other new users should copy your approach. (With 4x binning). I just have a few notes:
Are you taking only good 2D classes, or rejecting the clearly bad ones? I recommend the latter strategy; discarding junk is beneficial but rare views are likely to be in poor (but not junky) classes. Junky == not a (any) particle, half black/half white, bright white dot (usually little gold pieces breaking off into the ice), stripes/ripples (beam edge), etc.
Have you tried 3D classification (heterogeneous refinement)? I usually don’t use ab initio for choosing particle sets, instead I will start a het. refine using 1-2 copies of the best ab initio reconstruction, plus 2-6 alternate or bad (intentionally junky) ab inito volumes. You can also go back and use the particles from the first 2DCA selection here and potentially pull back in more particles than you can with multiple 2DCA rounds. If you have multiple conformations/compositions of interest, you can repeat the 3D classification on the selected sets of particles using just the good references. E.g. if you have mixed 80S and 60S particles you could start 3D classification with the 80S particles and the 80S and 60S references. Often you’ll pull out some additional misclassified particles this way.
These heterogeneous refinement steps are the only thing I do that differs significantly from your approach (I also tend to re-pick using templates based on my structures at some point). You can get very nice structures quickly and with little manual parameter optimization this way, but it does fall short for “resolution pushing.” So far only 3D classification in Relion with careful parameter choices has really helped me eke out a few additional tenths of an Ångstrom.
Just wanted to also chime in here and add to the excellent set of responses. It’s probably important to clarify/emphasize that 3D Classification and Heterogeneous Refinement are two separate jobs (not totally clear from Daniel’s comment) in cryoSPARC. What we call ‘3D Classification’ is effectively Heterogeneous Refinement but with the 3D orientation/ 2D shift of each particle fixed and some further improvements/differences in initial density generation.
In our testing, we’ve often found that Heterogeneous Refinement is better for identifying ‘larger’ density changes with a small number of classes, while 3D Classification can much more efficiently separate tens of classes, some of which may be useful. We’re actively working on improving the default parameters and optimization routine for ‘3D Classification’ so it can more reliably produce useful classes over sundry data to both identify heterogeneity and derive similar resolution improvements as RELION’s 3D classification routine.
on that point @vperetroukhin, having a touch more flexibility - not just classification without alignments, but also classification with tunable orientation searches (a “local” version of heterogeneous refinement) would be very helpful, for the cases in between the two that you describe.
We usually only use 2D for initial analysis & identifying subsets to use for ab initio reconstructions, then do heterogeneous refinement against the entire dataset with all the “good” models that have been identified via that route, as well as some junk decoys, either random density or specific (e.g. empty nanodisc/micelle classes).
Then after an initial consensus refinement (including local/global CTF if warranted), migrate to relion for polishing (sometimes it has a substantial effect, sometimes not). Repeat multiple cycles of polishing/refinement until no further improvements are noted in the consensus map, then proceed with further classification, local refinement, etc.
@olibclarke Do you have any opinion about how important the local motion estimates are to initialize polishing? We are going to add polishing-compatible output from motioncor2 directly. Starting polishing from whole-frame motion is trivial, converting the patch motion data is more work.
@DanielAsarnow not sure - haven’t really done the proper comparison. From the original Bayesian polishing paper:
“The particle trajectories for the Bayesian polishing were initialized with the motion estimated by our version of MotionCor2. This initialization does not appear to be strictly necessary, however, since in most cases the Bayesian polishing algorithm converged to the same optima if initialized with an unregularized global trajectory. On the β-galactosidase data set, for example, 90% of the final particle positions showed a difference of less than 10−4 pixels as a result of initialization.”
This would suggest that whole frame might be fine, but I haven’t really tested this myself. If you want a test case, try EMPIAR-10737 - this is a case where in our hands multiple rounds of polishing/CTF made a big difference.