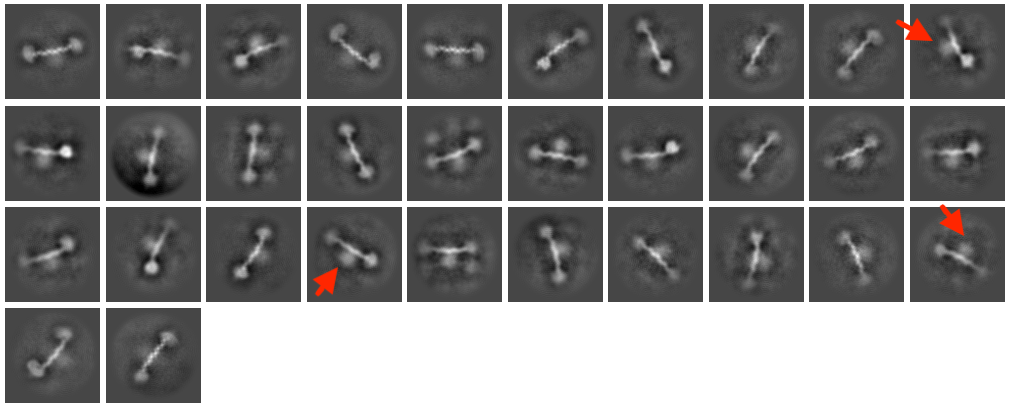
I am interested in improving the diffuse density on the middle of the DNA (marked by red arrows in the attached figure). The DNA has covalently linked proteins (~40 kDa) attached on either end to ensure the protein of interest (blurred density in the centre of DNA) doesn’t slide off.
Can anyone suggest a strategy to improve this blurred density? I have already tested different circular mask sizes in 2D classification with and without recentering, but that hasn’t been much help.
Any suggestions would be highly appreciated.
It’s always the part you’re interested in that doesn’t want to resolve…
It’s presumably blurred for one or two reasons:
It’s not there in the same orientation on each particle, so averaging them creates a blur
The orientation is ambiguous so the software itself can’t align it well.
#2 seems less likely here, given the shape the alignments should actually be a piece of cake, so I’d focus on #1. If it is problem #1, you are unlikely to solve it in 2D. You will probably need to take this structure to 3D and try 3D variability analysis with a focused mask around the central protein.
If the protein-DNA interaction is non-specific you may just be out of luck. It could be there in so many orientations that it will never resolve. But if it is there in only a handful of discrete orientations, it’s just plausible that the 2D classification can’t resolve it, but 3D variability might.
Getting this to a high-enough resolution structure that 3D variability works, though, is not going to be trivial, given the flexibility of the DNA.
Now that you have the blurry density picked and centered, reextract a box size that excludes the other structured portions (DNA) and rerun 2D or 3D with restrained motion (or recentering turned off). the Blurry part is blurry relative to the aligned poles. If they aren’t there, they won’t dominate the alignment (hypothesis - it’s also likely the DNA string dominates the alignment which would not be as lucky)
Have you played with the recenter mask threshold, number of iterations, and batchsize per class? I had a flexible portion of my protein that I was able to resolve decently once I played with these parameters (I was even able to resolve it into two separate classes by 2D). The “magic” numbers for me were: 0.4 for re-center mask threshold, 2 for the # of final full iterations, 40 for the # of online-EM iterations, and 1000 for the batchsize per class parameter.