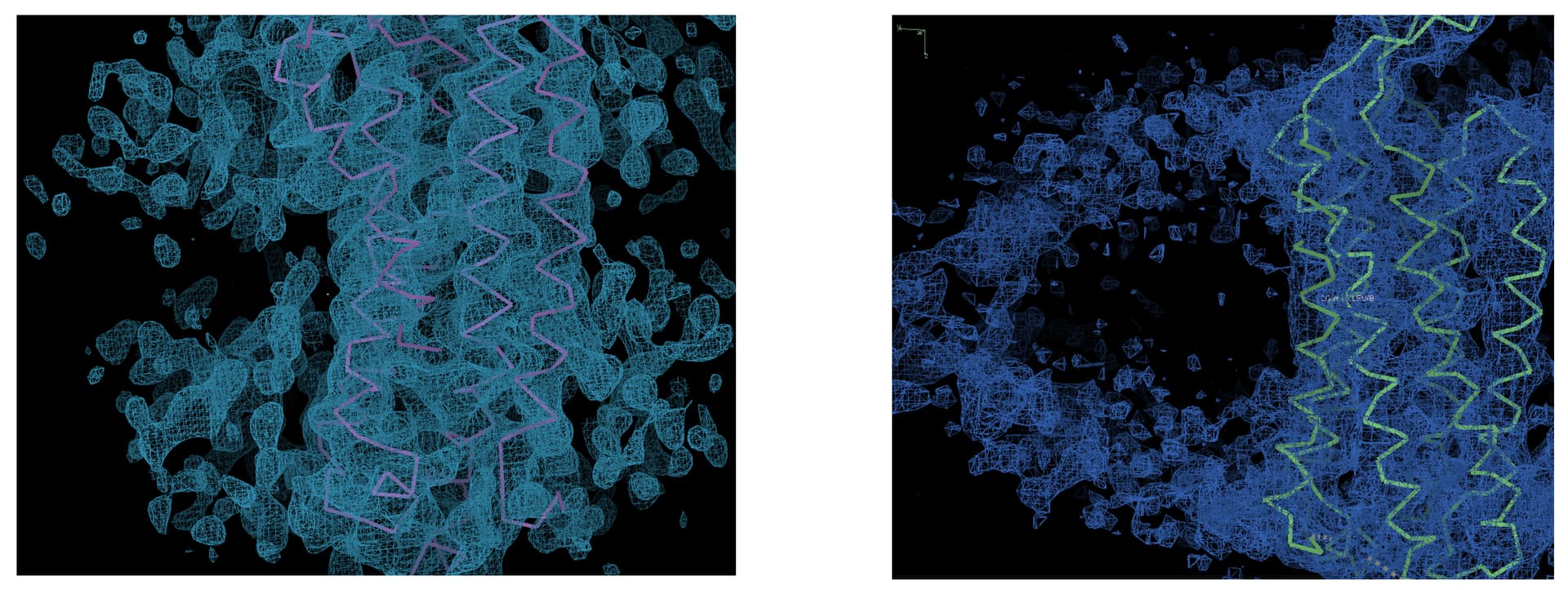
The two figures on the left and right show the transmembrane helix domain models and cryo-EM maps of two members from the same membrane protein family. The map on the left is the result of a local refinement in cryoSPARC targeting the transmembrane region, with a resolution of 3.9 Å. The map on the right is a composite map generated from cryoSPARC’s non-uniform refinement and local refinement, with the transmembrane region at a resolution of 4.3 Å, and it has been publicly published.
Why does the map on the left exhibit large, unexplained densities (contoured at 5.5 RMSD in COOT) near the sides of the transmembrane helices, while the structure on the right shows almost no such bulky densities near the transmembrane helices (the semi-circular densities are micelles, contoured at 3.0 RMSD in COOT)? How can cryoSPARC or other software be used to make the electron density map on the left as clean as the one on the right, free of large redundant densities?
Disclaimer: it’s tough to tell what’s going on from two static images.
But - this is a big problem, especially with membrane proteins and transmembrane regions of membrane proteins. Noise, vs resolution. The problem is that TM helices love to wiggle around. Detergent micelles love to get into the way of the mask, and cause mask edge artifacts. Particles love to orient upside down.
I have found that ignoring resolution, the map should be relatively noise free. Even a low res map should be obvious where the TM helices are. Where the cholesterol/lipids are (most proteins usually bring a few of this and they appear next to the TM helices as long blobs running parallel). Even if the resolution isn’t there to see side chains, if things are working, it should at least be clean.
There are a lot of problems with local refinement of TM helices. To be totally honest, it doesn’t really work. Maybe someone will correct me, but the mask edge artifacts you get of clipping into the micelle usually brings you straight back to the detergent blobology land. I’ve seen a lot of folks claim to have it working for GPCRs, but never really have I been convinced, when I look at the density it always looks like this - totally full of spurious artifacts we are supposed to ignore, and just see what our preconceived biases tell us.
The strategy I typically finds works best is just multiple iterations of 3D classification. I’m still not convinced whether this is finding particles that do indeed share the same 3D orientation, or rather just finding the ones that can computationally reinforce positively for whatever reason, but enough iterations almost always gets me to where I can get a clear, noise-free picture of where those helices are. You lose particles, so in theory, you lose resolution, but in practice, you wind up with a better understanding of your biological system.
Sorry that there’s no good answer, but yeah, what you are noticing is completely valid/legitimate and a real issue in this space IMO.
There are a lot of problems with local refinement of TM helices. To be totally honest, it doesn’t really work.
…is a little broad IMO - there are plenty of examples (e.g. in Architecture of the human erythrocyte ankyrin-1 complex - PubMed the density for Rh was lousy prior to local refinement, improved dramatically after; same true for band 3 & aquaporin) where local refinement improves density quality (not just resolution) within the membrane.
It does require careful parameter choice though - usually the default search ranges are much too large, and the initial lowpass too low res, and improvement should be assessed by visual interpretation of the maps, not solely based on FSC.
3D classification is definitely a complementary strategy - both have their place IMO
Sounds like you may want some postprocessing workflow: sharpening/density modification/map enhancement. These are aids for visualization not for automated refinement into and while useful for visualization purposes and as an aid in manually fitting still need to check multiple maps by eye.
To get rid of the density from the detergent belt, TM is wrapped with a belt but heterogenous interactions so get blobs, i tend to like EMReady. Havent used EMReady in scipion (so unsure how easy that would be to install for plugin) but a conda environment of EMReady works on a number of our machines. Its a density modification program, will completely remove the detergent belt leaving the TMs. Depending on the lipid density some stable lipids will still be present after processing. EMReady has its caveats, low density areas likely to disappear (hence the removal of detergent belt) and it can at times connect things that probably shouldn’t be connected (salt bridges).
There are multiple other programs out there to try: cryosparc has deepEMhancer, phenix.resolve_cryoEM, locscale, cryoSAMU are just a few.
Local refinements should definitely improve the quality of the map significantly, we have many datasets where this is the case. Tightening up the search parameters is definitely the way to go like Oli mentioned.
If the density still looks bad, there is probably significant heterogeneity in the dataset. I would try extensive heterorefine with maps representing the heterogeneity generated with ab initio. 3D classification we find is really only useful once the data is very clean and aligning well in local refines. Even still, I would prefer relion class3D because it will give more control. Hope this helps.
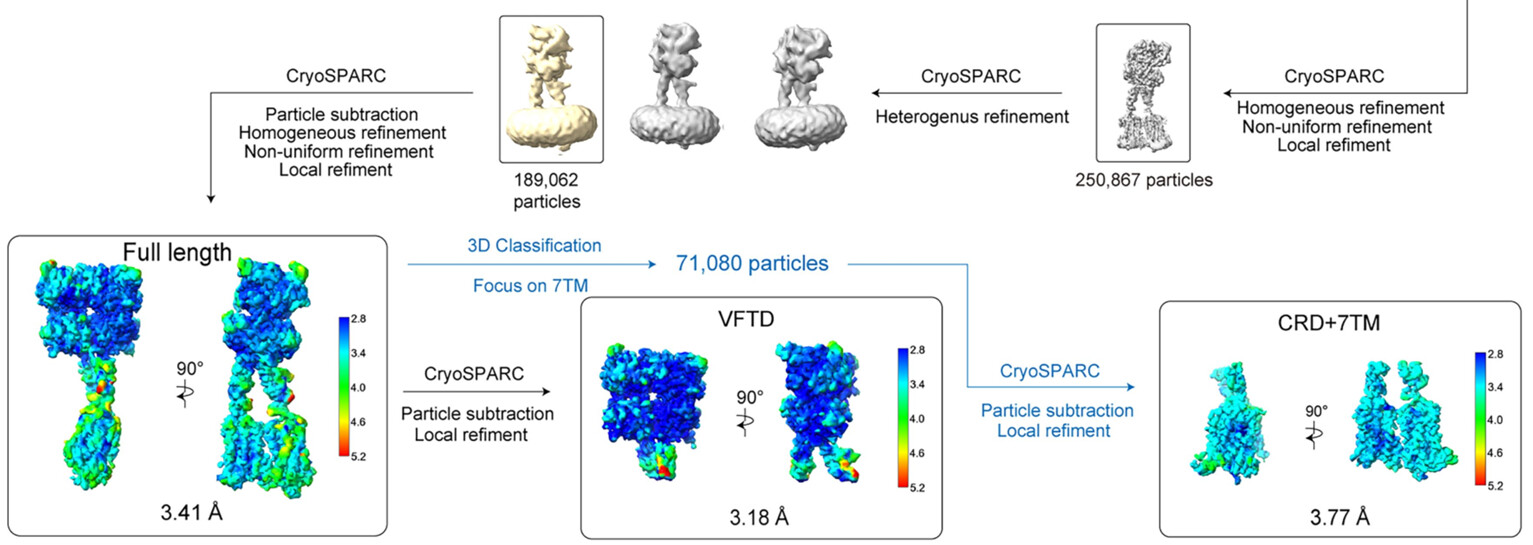
You are right, careful parameter choice will be needed. However, after many trials, I don’t know on which point the map quality will converge, and if we should collect more data or should try more choice on parameters for local refinement and mask, such as dilation radius, soft padding width and search range. The 2025 NATURE paper on sweet taste receptor showed a very clean map on 7TM. Can you analyze their parameter choice or give some comments on their mage data collection that likely led to such a clean map?