Dear cryoSPARC community
I am encountering difficulties with refinement of my filamentous protein.
I am processing based on the recommendations of this guide:
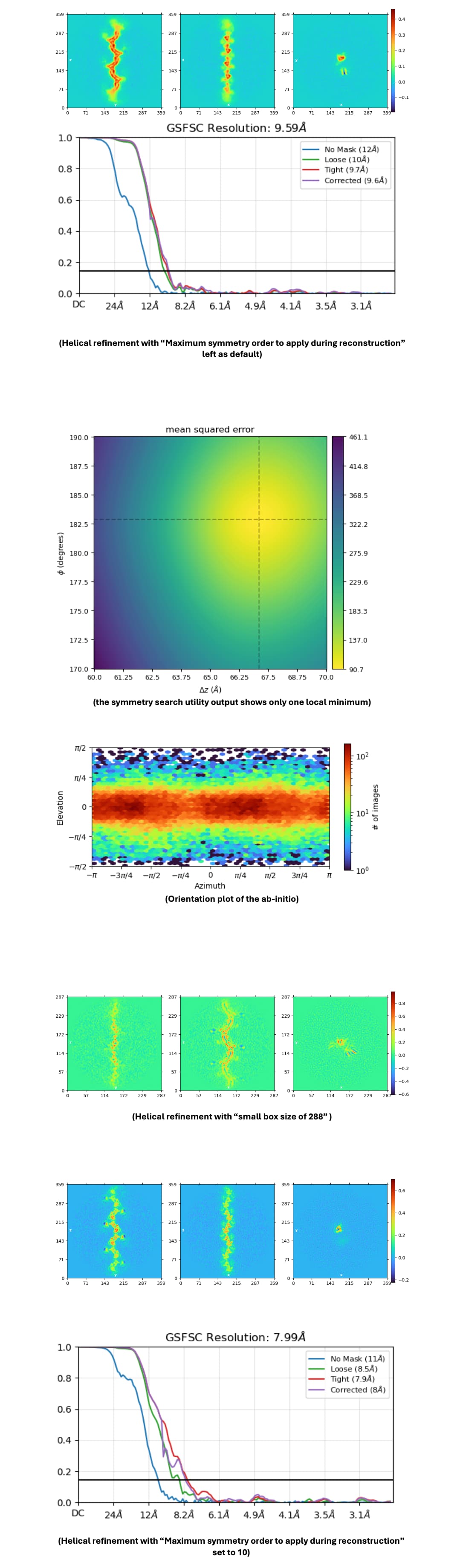
This is a dataset based on ~72 000 particles, obtained from different collection sessions (sharing the same collection parameters) picked with the filament tracer. Particles are picked every 67 A, based on the helical rise obtained from symmetry search utility. It is a flat filament with a helical twist of ~180 degrees.
Running the the symmetry search utility on the ab-initio finds only one local minimum, so i do not expect any problems there.
Based on the ab-initio, it does not look like there are any orientations missing.
Yet when i run helical refinement, I do not get the resolution I expect for this dataset, only around 8 A
I have tried a smaller box size, which led to a slight improvement of resolution but overfitting in the model.
I have tried increasing the “Maximum symmetry order to apply during reconstruction” to 10, only resulting in marginal improvement.
Do you have recommendations for refining such a protein?
Or should i include more detail in this thread?
thank you in advance!
Sven