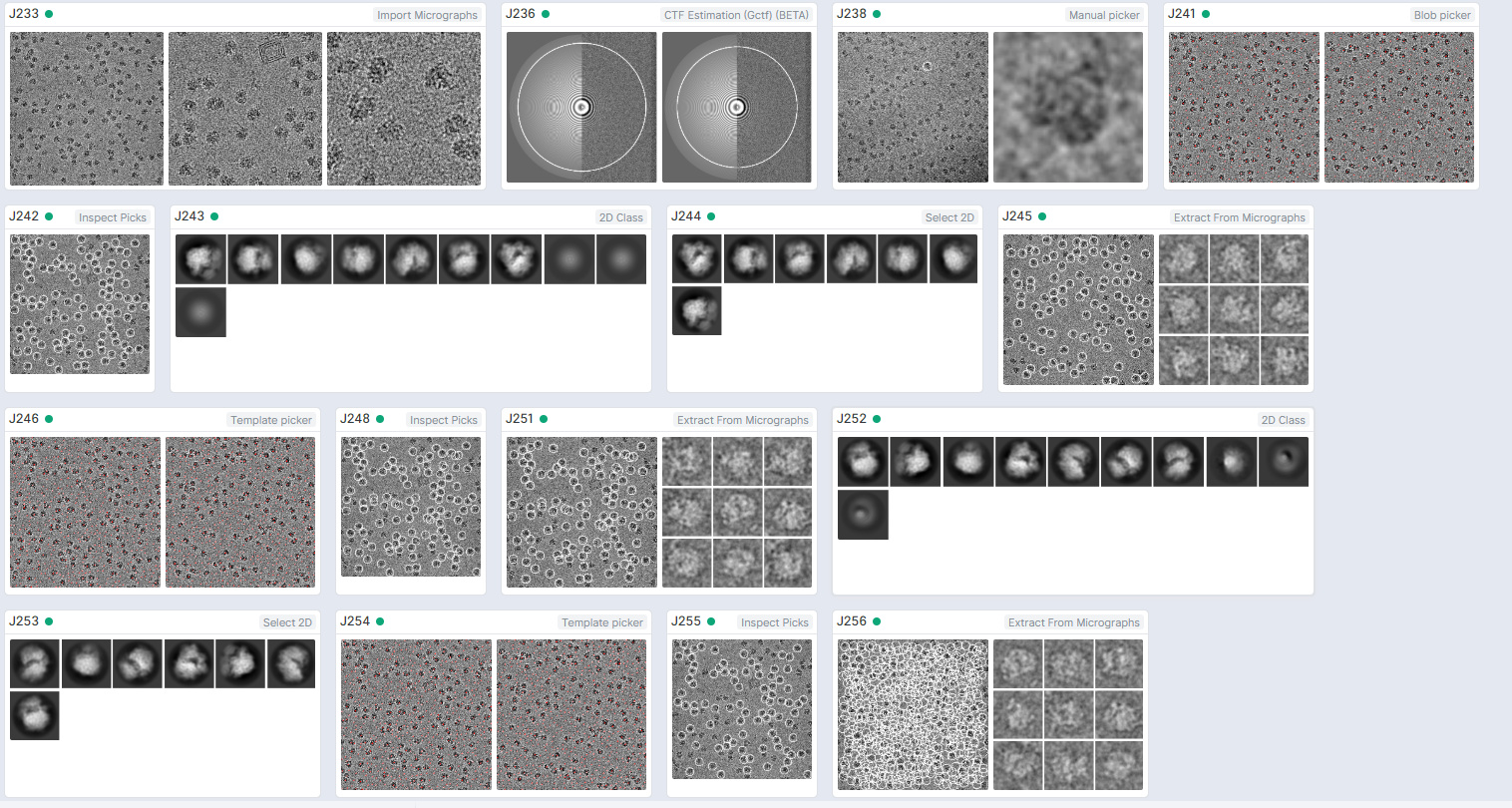
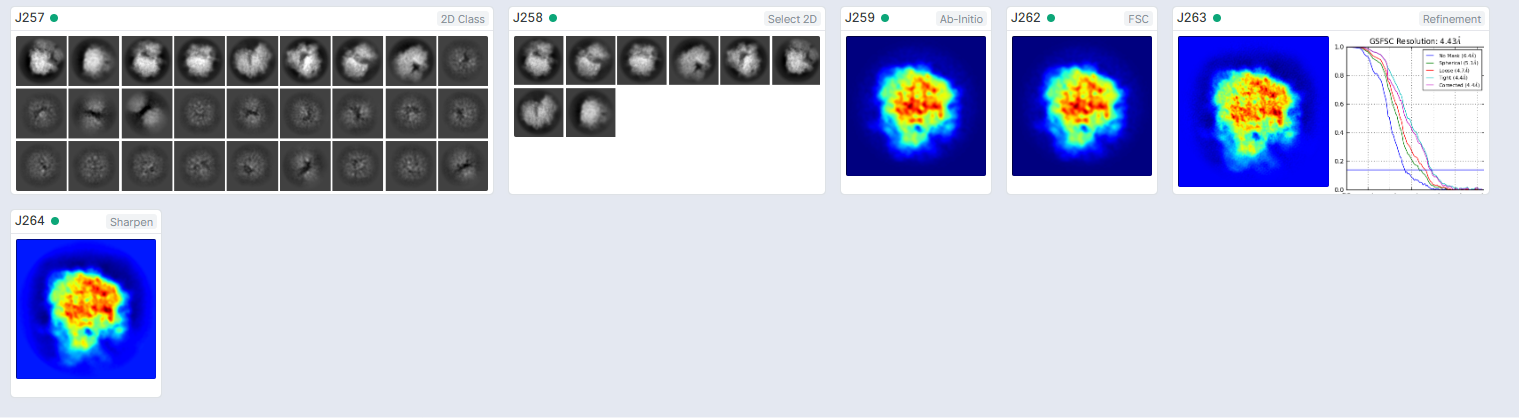
Hi,
I am using ribosome data from the EMPIAR data bank. The published results have <3A resolution. However, i get >4A at best when I use hetero/homogenerous refinements. Is there (in general) any way to make the reconstruction sharper, that wasn’t mentioned in the tutorial workflow? are there limitations to the software?
I would appreciate if you help me with this issue.
Stay well.
You will probably need to provide a bit more info about your workflow and the dataset to get helpful responses. It is certainly possible to get high resolution reconstructions using cryosparc, and I would be very surprised if you could not do so for a ribosome dataset.
The global resolution you can achieve with cryosparc should not be that much worse than published.
I would not be surprised if sorting will not reveal the same conformational states but this is no matter of resolution.
Did you check pixel size and ctf parameters?
What is your workflow - what jobs did you use in cryoSPARC, did you start from the same number of micrographs as the authors, are the number of picked and final particles comparable to what the authors originally obtained, etc - more detail needed to figure out where things are going wrong
your box is too small. for a pixelsize of 1.34 I would use at least 300 pix.
Think about starting with binning and sort out heterogeneity by heterogeneous refinement.
Try ctffind4 with default parameters.
Take care the CS of the Polara is 2.0, not 2.7 as for Krios.
Yes - box size way too small, and I would also use many more 2D classes than what you seem to here.
I would suggest initially binning to ~4Å per pixel, and then re-extracting after 2D/3D cleanup. I would also suggest the use of Patch CTF, which in my experience does a better job than Gctf.
Also why are you using so few micrographs? The fact you are using less than half the micrographs, but are ending up with ~3/4 the particles would suggest you haven’t cleaned up enough yet.
You are also importing the aligned sums, rather than the movies from what I can see, so per particle motion correction is not available to you, making it difficult to compare to the published structure which was obtained after polishing.
Good luck, looks like a good dataset to practice on, hope this helps!
Hi, Thanks a lot for your suggestions. Which job is related to binning?
(in which job do I set it) and what is the criterion for binning? Thanks again.
Hi Arya, you can bin particles during extraction - e.g. if you extract with a box size of 400 and resample to 100, that would corrrespond to 4x binning, or binning to 5.36 Å/pixel. You can also bin using the Downsample particles tool.
@Arya please do update on your progress if possible. You should be able to easily get 3A or better on this dataset when using all the micrographs/particles.