Dear All,
In the local refinement job, I noticed that FSC-mask auto-tightening makes the resolution substantially worse.
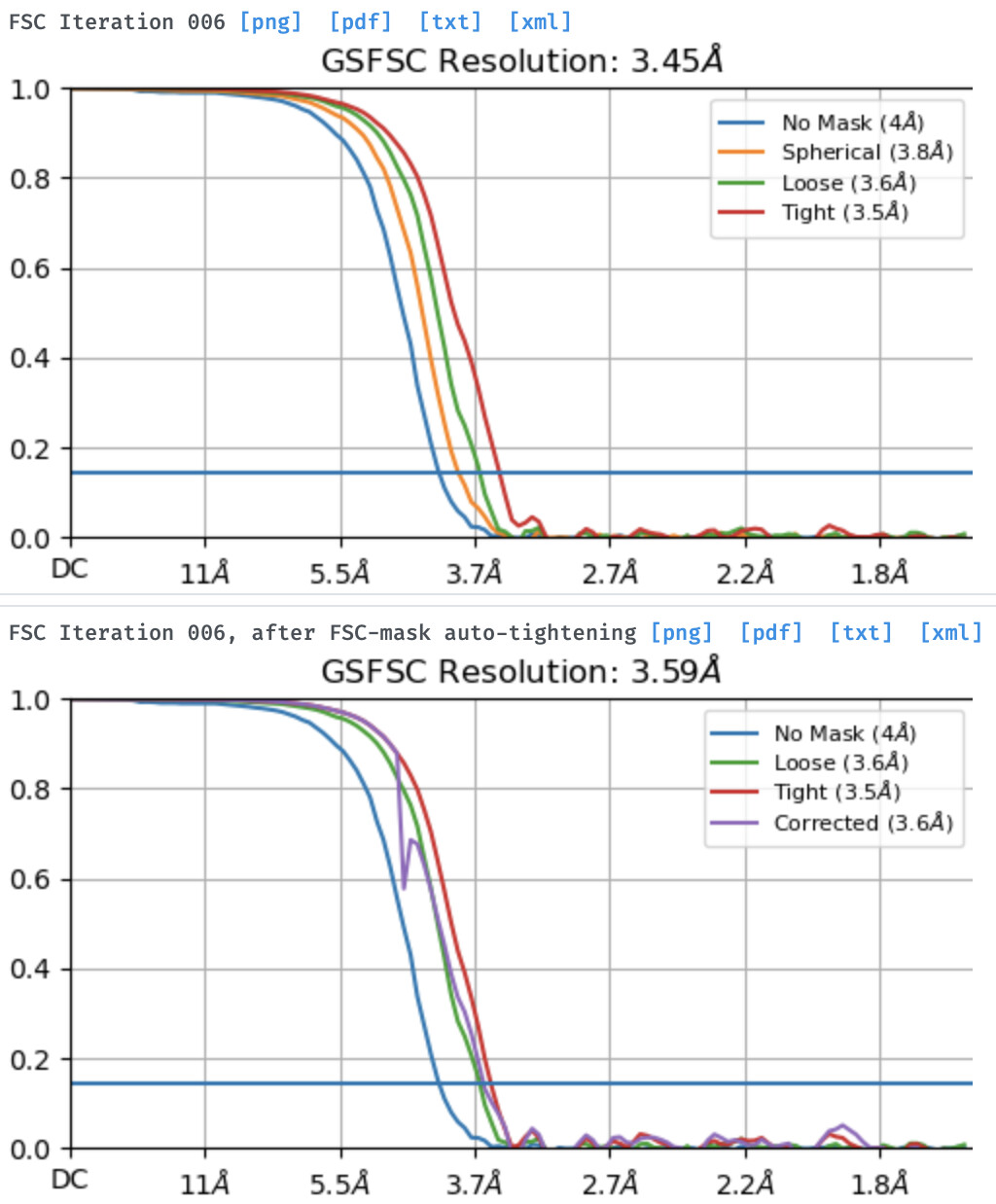
I am wondering if the output maps, in this case, are from the refinement before mask auto-tightening at 3.45 A or from after mask auto-tightening at 3.59 A. Any suggestions to improve the local refinement job are greatly appreciated.
I am using cryoSPARC v3.3.1+220118. This is an HIV Env trimer in complex with an antibody. The overall resolution is ~2.85 A. While the density for the trimer is very clear, the density for the antibody is very poor. I can see density for only the parts close to the trimer; the constant domains and part of the variable domains away from the trimer are without density. For the mask, I docked the antibody variable domains in the density and use molmap in Chimera to create a volume of the antibody variable domains. For the alignment parameters, I used standard deviation (deg) of prior over rotation 5, standard deviation (A) of prior over shifts 3, and turned on both re-center rotations each iteration and re-center shifts each iteration. For the refinement parameters, I used number of extra final passes 3, and initial lowpass resolution (A) 6. Using default parameters gave resolutions of 3.77 and 3.89 A before and after mask auto-tightening, respectively, and the output map is very noisy.
I am new to cry-EM, and this is my first attempt to do local refinement. Please give me suggestions in as much details as possible, as I am still trying to find my ways around. I greatly appreciate any suggestions to improve the local refinement results.
Best,
Shuishu