Dear
Recently, I got a sample that could form the filament. I meet a few questions.
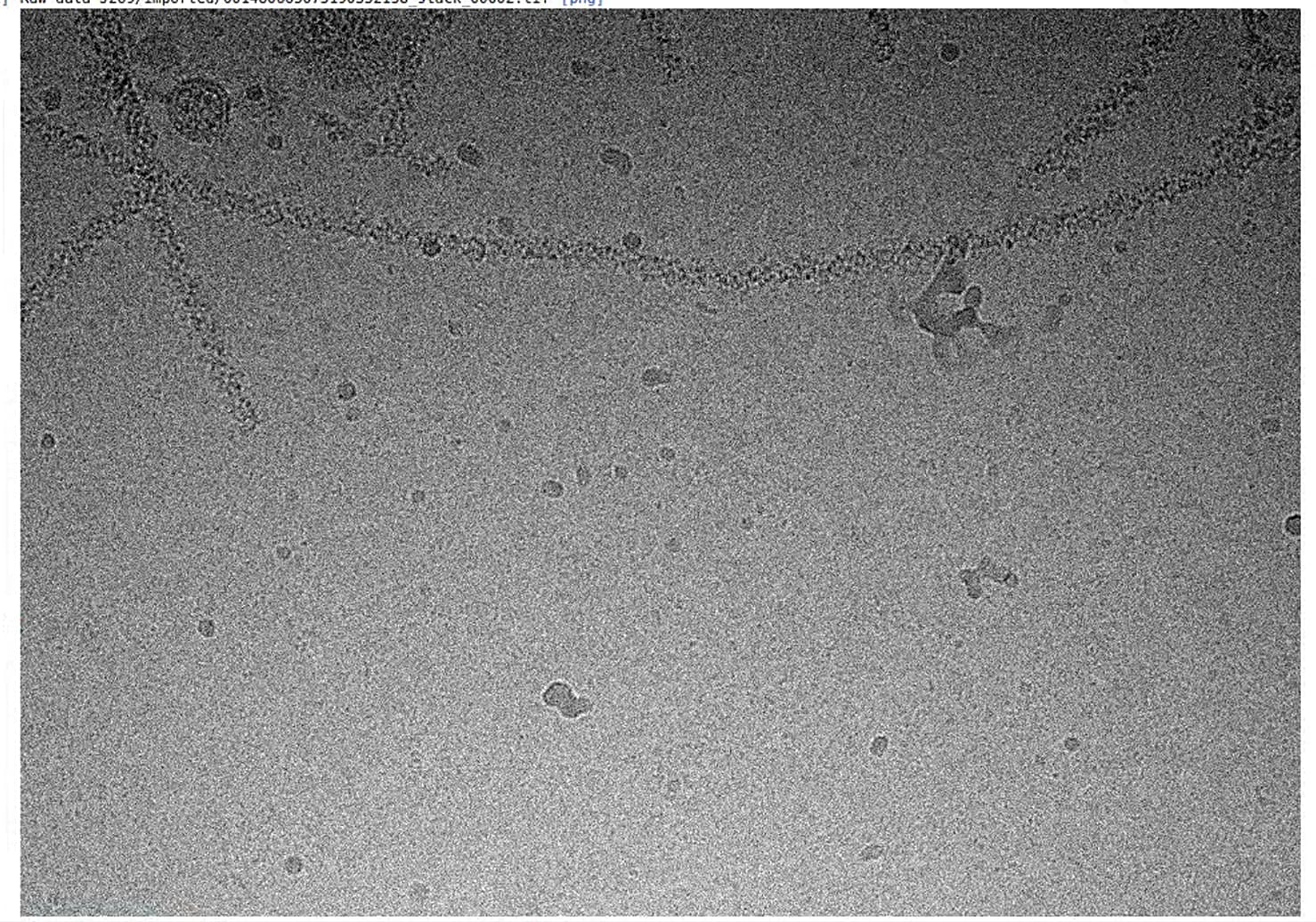
- I treated it as the common sample cause it was not filled with the images(shown in Figure 1, I got 3,000 movies in total). I didn’t use the filament tracer. I just chose a normal routine and it did work. However, the box size was the point that confused me for a long time. 256,320,384,640, I tried all. But none of them is good. The 2D classification looked good but the final resolution is just 6A(Fig2&3). So, which size should I choose?
- When I use the different box sizes and min. separation distance, I found some parts were picked more than one time and I am not sure that would have an impact on the final result AND I am not sure if should i use “remove the replicated particles” or not.
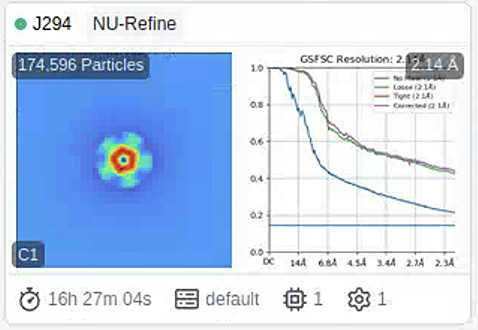
- The refinement problem. I found it could be the highest resolution in theory (the pixel is 1.067, and the shown is 2.14A, as Fig4). But when I downloaded the map, it showed very poor resolution. So, any way to avoid this?


And any suggestion is welcome! Thank you very much
Best
quwujian
1 Like
You absolutely should. That FSC curve is showing the classic signs of duplicate particles.
Helical reconstruction is tricky, and a lot of care must be taken to avoid this issue. If you follow a processing pipeline designed for helical work (filament picking, etc) it has some safeguards. 
If you remove duplicates, I suspect you’ll see the FSC curve drop to zero around 6 Angstrom.
The CryoSPARC guide has some guidance regarding helical reconstruction.
1 Like
Thank you for your reply. I will try it. But I am also confusing why it is hard to get the high resolution. Not enough particles? Need I should collect more images? Or preferred orientation problem? Thank you again.
With correctly applied helical symmetry, if the sample isn’t contaminated or heterogeneous, you’ll probably find that the resolution improves dramatically.
Preferred orientation isn’t really an issue with helical processing because of the symmetry application.  The guide covers how to estimate the parameters necessary to apply, and then how to optimise them. But local minima are a real danger in helical work, so be cautious and best of luck with your processing!
The guide covers how to estimate the parameters necessary to apply, and then how to optimise them. But local minima are a real danger in helical work, so be cautious and best of luck with your processing!
Perhaps the CryoSPARC helical processing tutorial (a link is on the page I linked above) would be a good idea to run through? There’s lots of extra things to be careful about with helical processing beyond the normal concerns of SPA.
2 Likes
Well, thank you. The sample is fine. I have built the model based on one map, so I am sure there is no contamination. I tried D2 symmetry before and it did be better before. However, our lab member suggested not using symmetry easily. Now I will follow your advice, read the tutorial and try it. Thank you very much.
1 Like