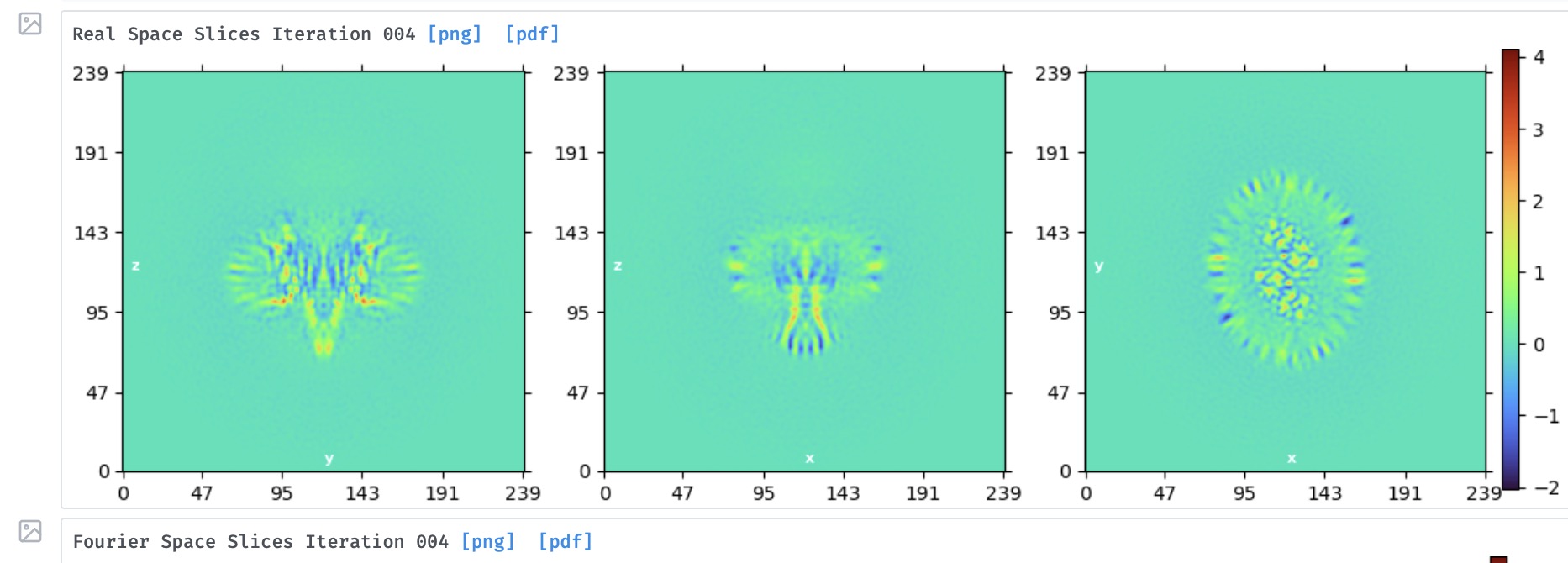
I‘m processing a membrane protein with c2 symmetry (120kda), here shows a local refinement results, so far I have got a relatively high resolution map but lacking a good secondary structure, I have tried to filter my datasets through 3D classification, hetero-refinement and Abi-initial, but all have similar results. So my question is whether there is a dominance orientation problem in figure 2.
Any suggestion to improve data processing are welcomed, thanks!
The orientation plot does not suggest preferred orientation. The map has severe overfitting streaks near the edge of the micelle - this often suggests that a lot of non-particles or junk are remaining in the dataset. How many particles do you have remaining after classification?
Thanks! Oli. I actually tried ~140k or ~14k particles after classification and both maps had similar issues (overfitting, figure 1).
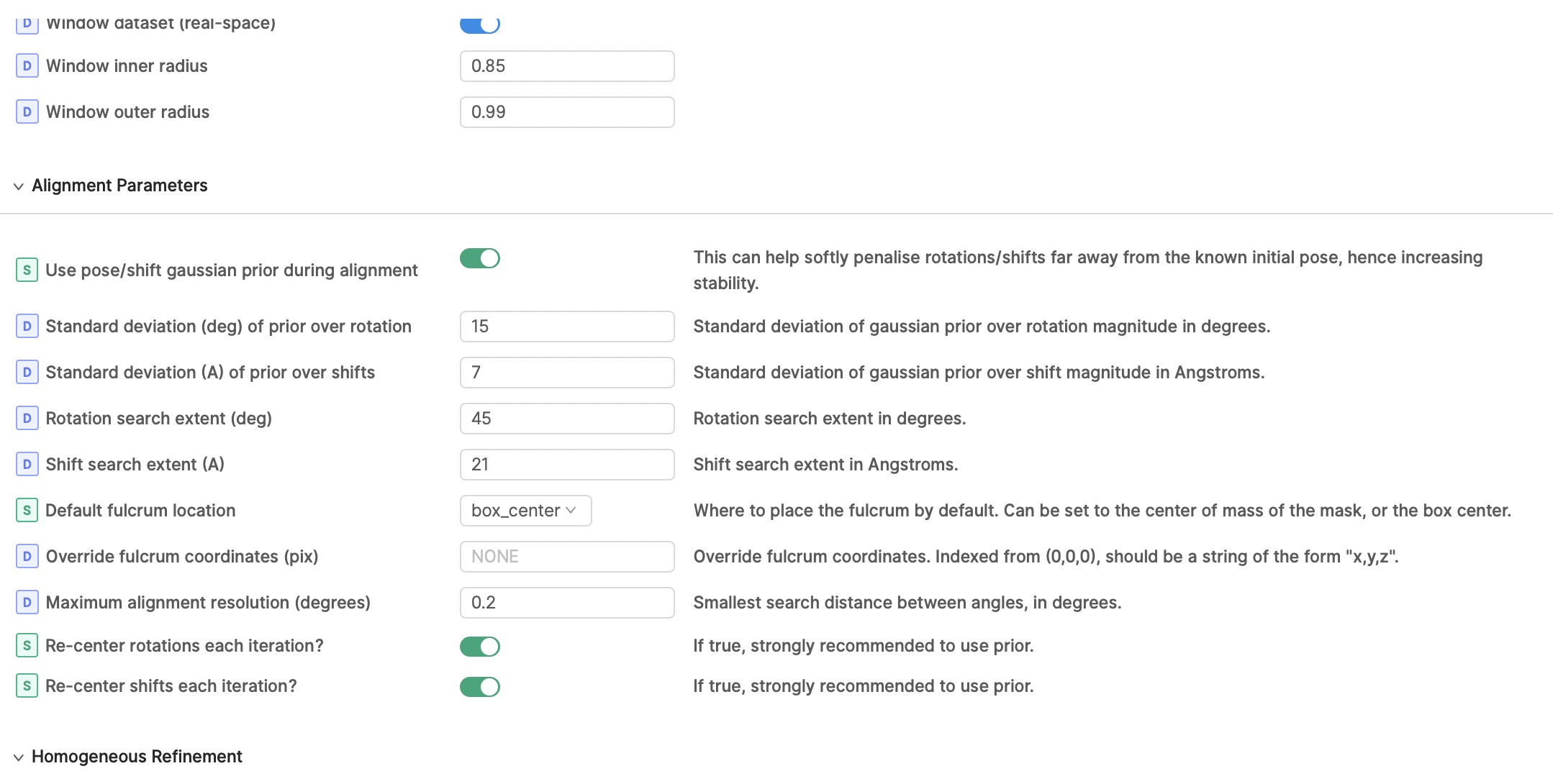
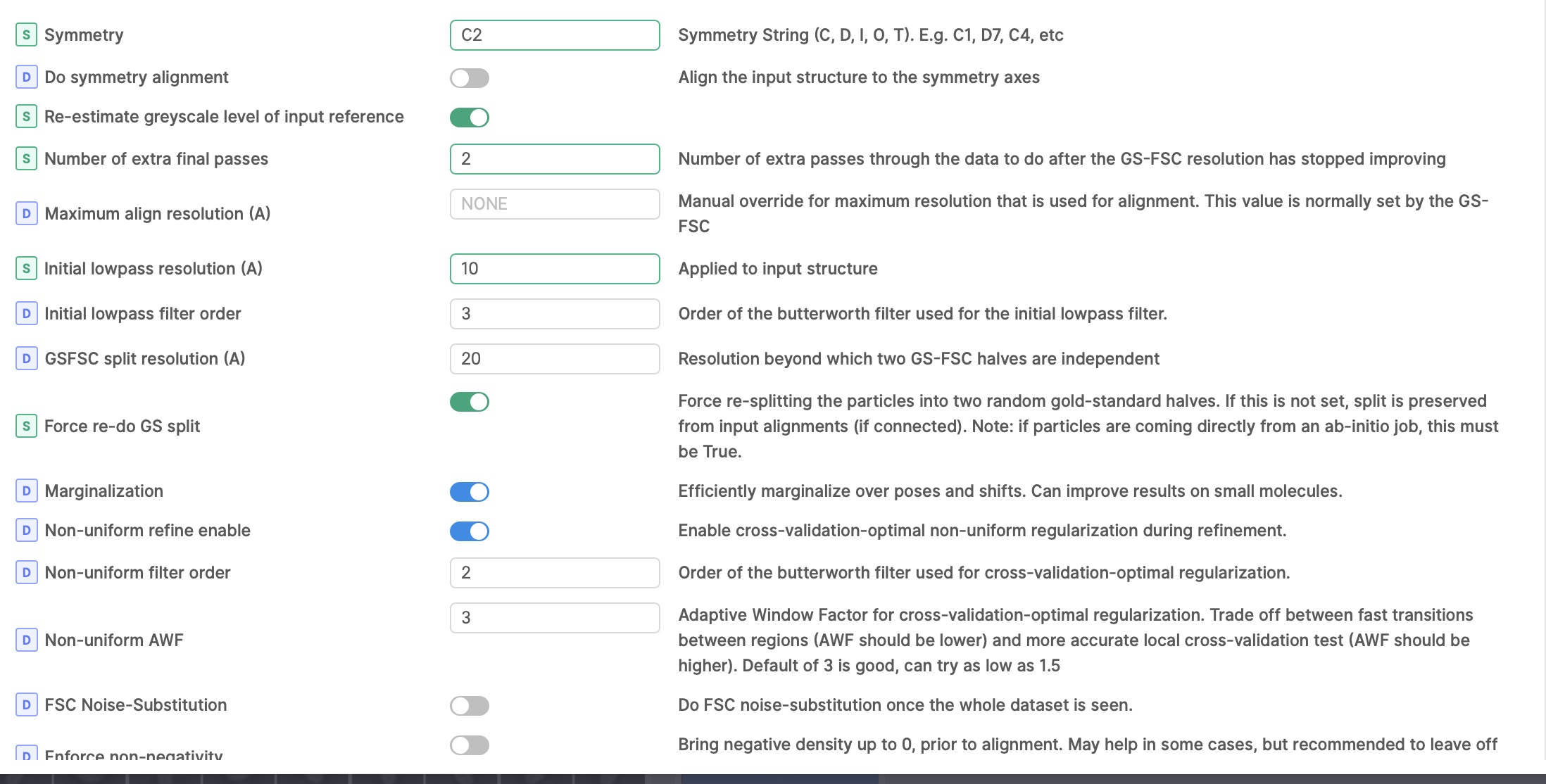
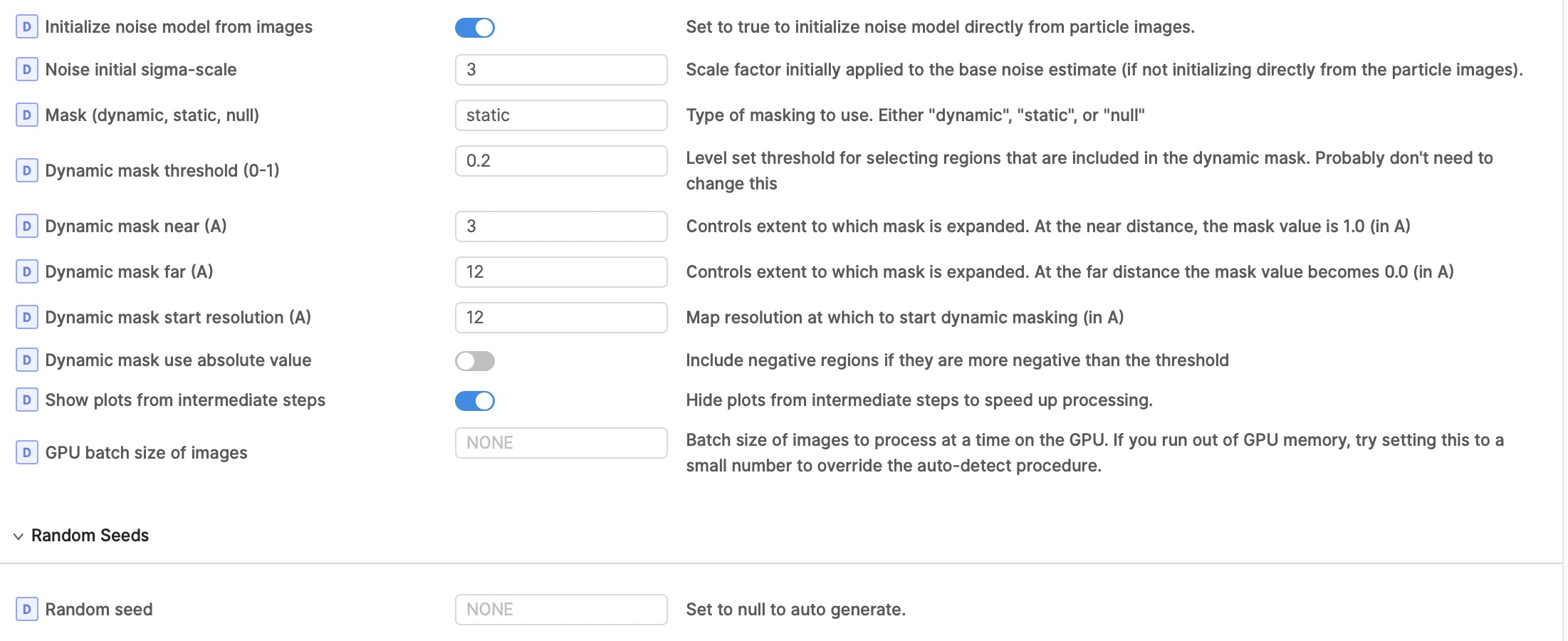
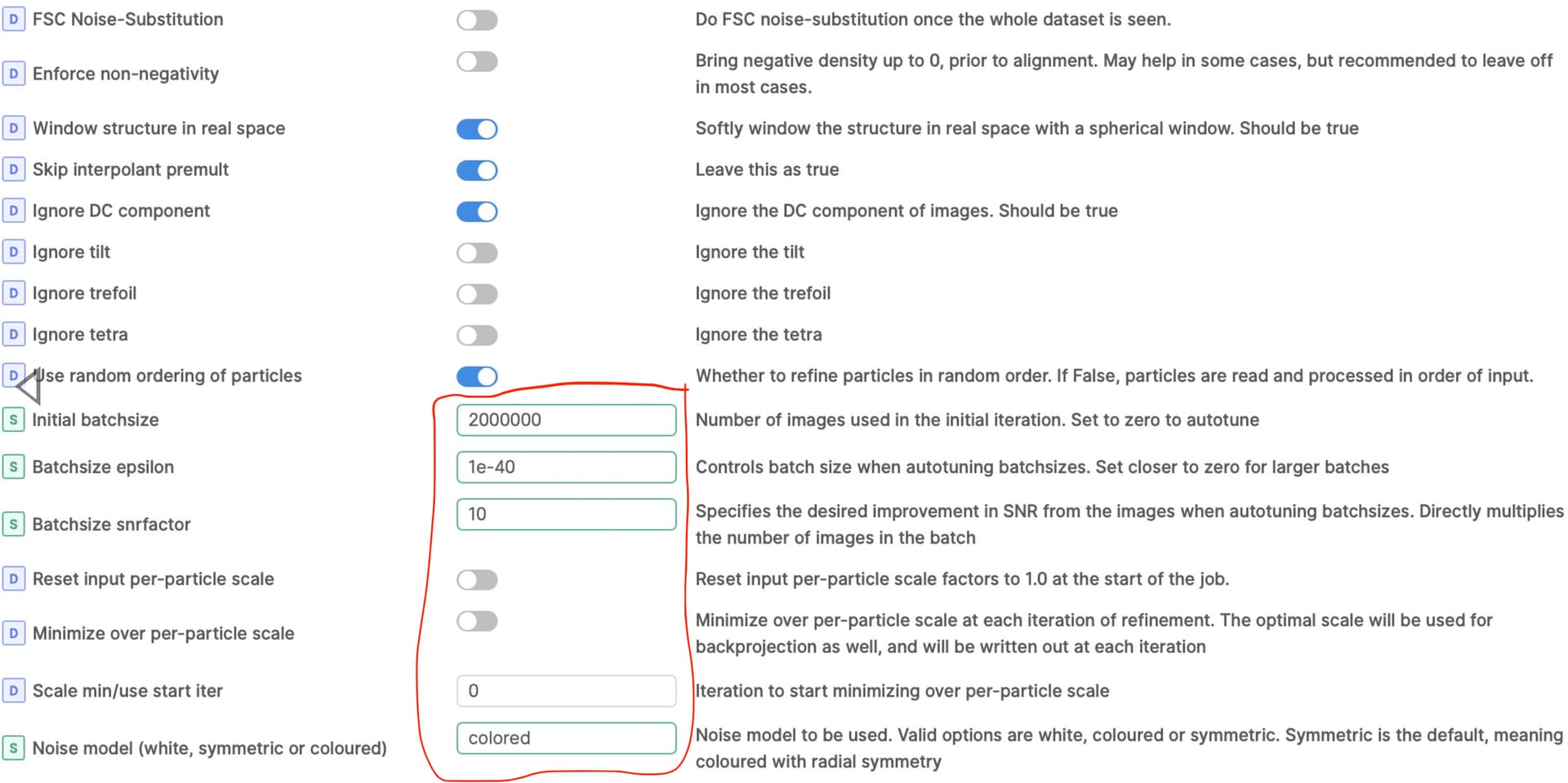
I also thought there might be junk particles in my dataset, I’m curious why I can’t discard it with 3D classification? I also run 2D classification with 140k or 14k particles, but both are pretty good. So do you have any suggestions for classification or refinement parameters? (following show my local refinement parameters setting, figure 2-4
Also, the search parameters you are using will likely give significant overfitting for a small membrane protein - I would start with much smaller search range and priors, and not recenter rotations and shifts each iteration. I would suggest priors of 6/2 and search range of 18/6 as a starting point.
How did the preceding global refinement look?
Are you sure it is actually C2, and how did you confirm this?