Hi,
I’m trying to solve the structure of a small protein-RNA complex (~60 kDa, RNA is a 26-mer oligonucleotide). I collected ~15K movies on a TFS Krios G4 equipped with an energy filter (70 e-/Å2 dose, 0.5-2.5 μm defocus, 0.47 Å/px, 10 eV slit width, Falcon 4i camera). I manually sampled ~2K exposures with good optical properties after patch motion correction to run a preliminary reconstruction. As the structure of the apo-protein was known beforehand, I used a 20 Å low pass-filtered template generated from the atomic model to automatically select particles. I manually filtered the particle picks to a total of ~1.5M particles out of ~2M picked by the algorithm and used ~800K particles to run 2D classification.
Unfortunately, I invariably got junk-like 2D classes only regardless of the alignment parameters used. Below are the results from a few example runs.
I’ve attempted to (1) limit alignment resolution to 6-9 Å, (2) change no. of 2D classes, (3) lower re-center mask threshold to 0.05-0.1, (4) increase online EM iterations to 40-100, (5) increase batchsize per class to 200, (6) turn off force max over poses/shifts, and (7) use clamp solvent. None of the attempts or their combinations have been helpful so far 
The particles show a very low SNR in the micrographs and look a bit amorphous (especially at high defocus values), but looking at the simulated TEM images of the apo-protein (generated using the Simulate Data job) I still think that I do have the correct particles distributed in various orientations. (Perhaps patch motion correction was not successful?)
(I’m using CryoSPARC v4.4.1)
What do you guys suggest I could do to try and get better alignment results? Any comments or suggestions are greatly appreciated 
Thanks in advance!
Sincerely,
Joonyoung
2 Likes
Hi Joonyoung,
What box size are you using for extraction?
Also, how thick is your ice? Do you have a power spectrum of a micrograph without gold/carbon?
Cheers
Oli
Dear Oli,
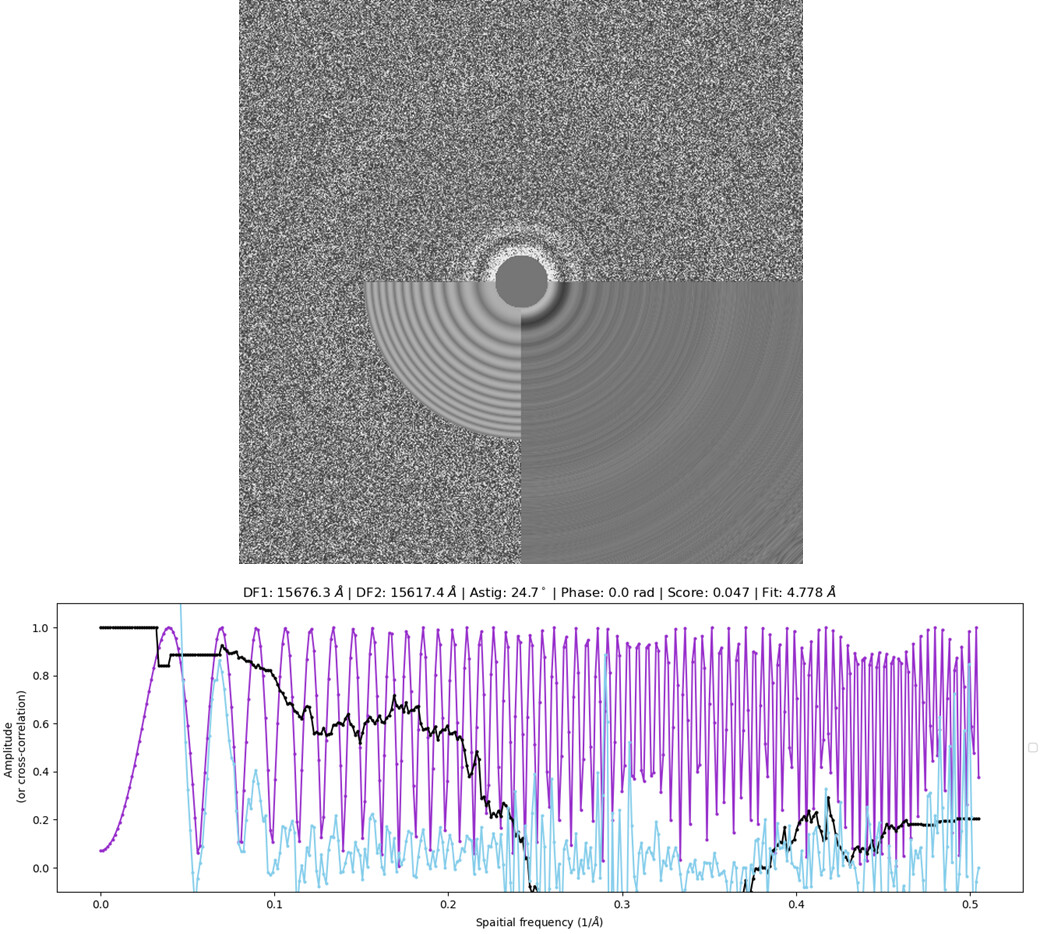
I’m using a 300 px box size (without binning) for extraction. While I don’t have the exact ice thickness in terms of nm, I have observed that the particle morphology is roughly the same in regions with thin & thick ice as judged by the EPU ice filter. I have also tested different blot times (3-6s) during grid screening but obtained similar results.
Below is a typical power spectrum and CTF fit found in my dataset (CTFFIND4).
Thanks for your interest!
Sincerely,
Joonyoung
Hi Joonyoung,
300px (141 Å) may be a bit small when CTF delocalization is taken into account at defocus values of ~1.5µm. Have you tried a larger box, with binning? Say 512px, binned to 64px?
Cheers
Oli
1 Like
Dear Oli,
I’ll try that out and see what happens  I’ll post an update once I get the results!
I’ll post an update once I get the results!
Thanks for your suggestion!!
Sincerely,
Joonyoung
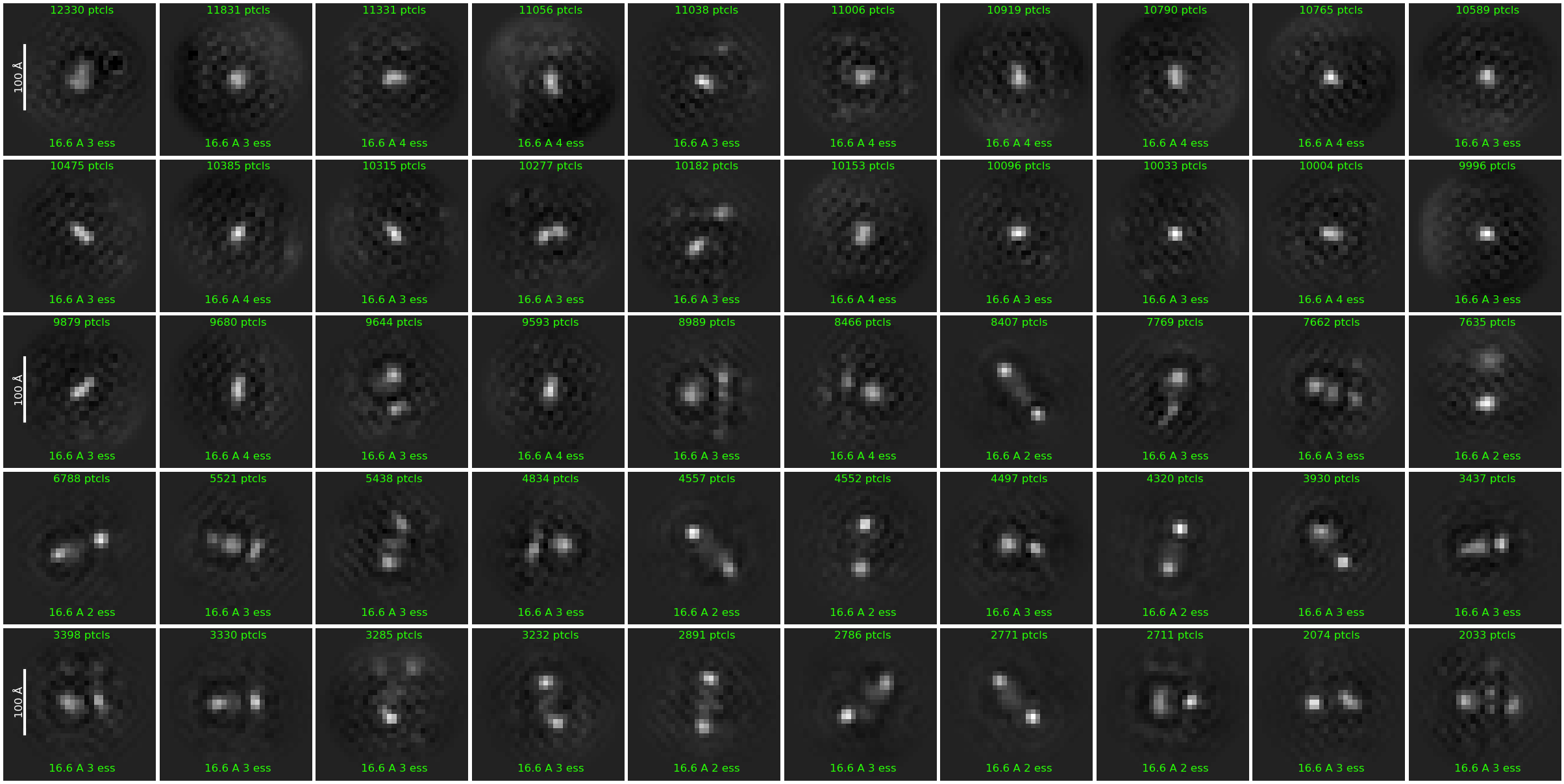
As per Oli’s suggestion, I increased the extraction box size to 512 px (F-crop to 256 px) after template picking. I also ran 2D classification in two steps: (1) Initial classification limited to 18A resolution to weed out obvious junk and (2) secondary classification (6A limit) with a 70A circular mask.
-
Initial 2D alignment
-
Select classes from step 1
-
Secondary classification
The 2D quality improved significantly, and I believe I can now recognize the overall shape of the complex consistent with the known apo structure.
The classes however still look a bit noisy and lack secondary structure details. What would be something I could try to perhaps improve the alignment quality even more?
Thanks a lot!
P.S. Great thanks to Oli for his suggestion regarding the extraction box size
Sincerely,
Joonyoung
1 Like
Some of these (e.g. below) look pretty nice now! Agreed the background is still a bit noisy. Sometimes increasing the number of full iterations at the end of classification to 10 or even 20 helps with this, in difficult cases.
I would also try just taking the decent looking classes from this classification and proceeding to multi class ab initio, and seeing what you get?
Cheers
OIi
Dear Oli,
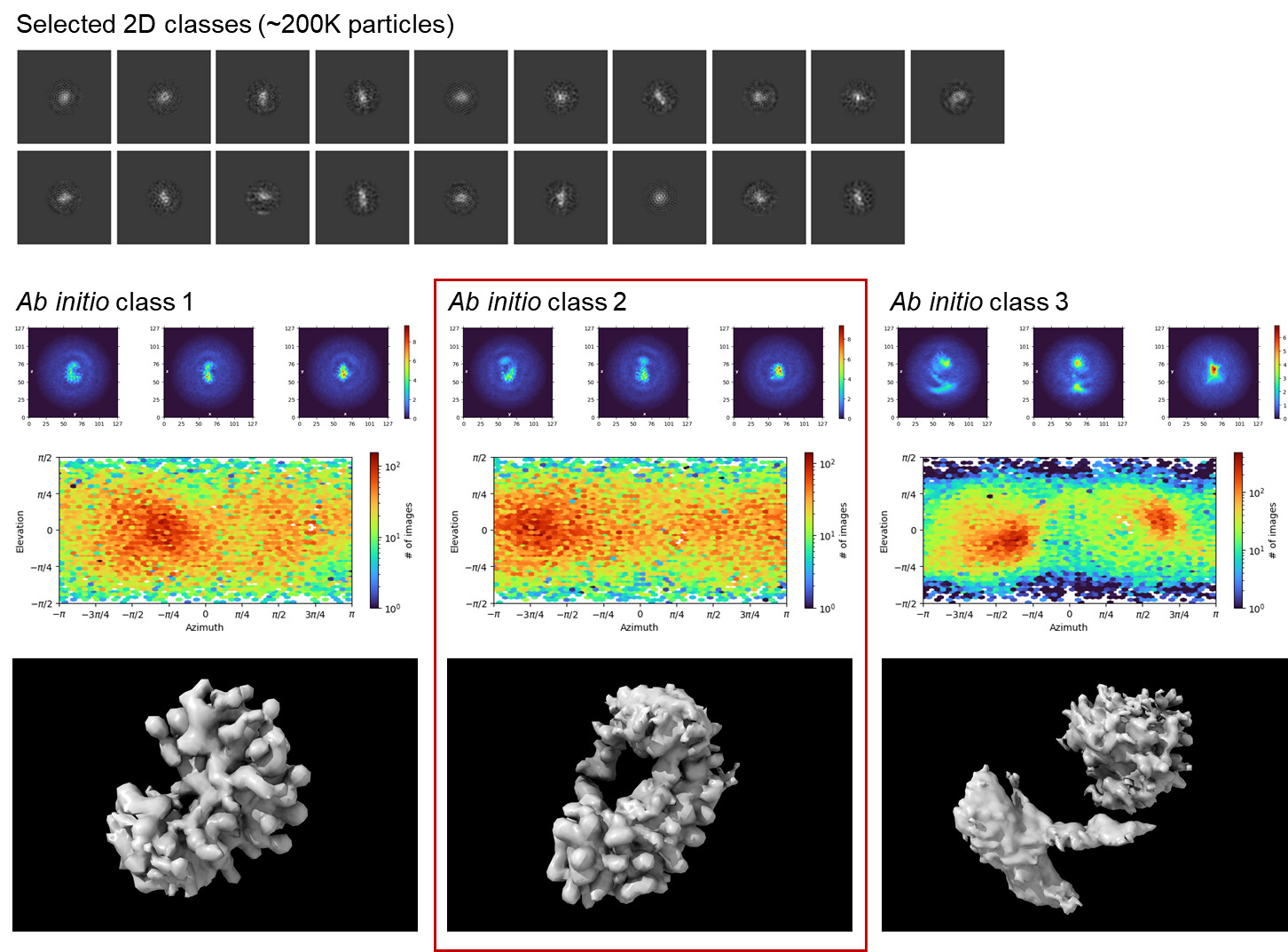
Thanks for your suggestions! I re-extracted ~1M high-quality particles from the micrographs and ran two rounds (18Å-limited → 6Å-limited) of 2D classification as before. While the class averages were still kind of blurry (even with added iterations), I was surprised to get reasonable-looking ab initio volumes in a 3-class run. Class #2 in particular was very promising as it showed good agreement with the known apo structure of the protein component (I unfortunately cannot disclose the overlaid image on the public forum due to community guidelines though I’d love to share it ). I am truly excited to finally see some good protein-looking densities in my reconstruction!
NU-refinement on ab initio class #2 yielded a 7.1Å map that looks consistent with the reported GSFSC resolution although the B-factor seems a bit unreasonably large. Hopefully the tube-like densities are actual secondary structure elements rather than an overfitting artifact.
I’m currently trying to improve the resolution by playing around with a few NU-refinement parameters. Regardless, this is a major jump from the featureless class averages I had just a week or two ago. Thank you @olibclarke for your invaluable contributions to this project! I’ll be sure to give a shout-out to the CryoSPARC community once this structure gets published 
Sincerely,
Joonyoung
Looks like progress!
I think there are some mask-edge overfitting artefacts in your NU-refine job (common for small particles).
I would try disabling dynamic masking, by setting the resolution to start dynamic masking to 1 Å, and maybe starting the refinement with an initial lowpass of 15 rather than 30 Å if the ab initios look decent.
Cheers
Oli
2 Likes
Dear Oli,
Indeed you are right. I see spike-like densities on the surface (not consistent with the expected fold of the protein) that indicate overfitting. Local refinement w/ gaussian prior ON using a loose static mask generated from the ab initio volume appears to yield better results compared to NU-refine. As you suggested, increasing the initial lowpass resolution does tend to improve the map quality as well.
Unfortunately, the resolution is still stuck at 7-8Å even after global/local CTF correction. I currently have ~65K good particles left after multiple rounds of 2D/3D cleaning (from 10M(!) initial picks). With such a small particle stack I don’t know if I could afford to further remove junk (as is usually attempted to improve resolution) without compromising the theoretically achievable resolution (as per the B factor). It seems that the low yield of “good” particles in Cryo-EM is a universal phenomenon for small (<100 kDa) particles as reported in this 2019 Nat. Commun. paper.
A notable commonality that has emerged from the high resolution structure determination of many different types of samples by the cryo-EM community is that relatively few (e.g. <20%) of the initially picked “particles” are retained in the final high resolution reconstruction(s). This trend seems to be particularly true for the smaller macromolecules presented here, with only ~2% of the ADH or ~7% of metHb particles selected from 2D classification contributing to the final reconstructions.
What might be something I could try to potentially push the resolution up into the 3-5Å range with the current dataset?
Thanks as always and merry christmas!!
Sincerely,
Joonyoung
Hi Joonyoung,
Re-reading your initial post:
I would be careful doing this. While there is nothing intrinsically wrong with using model-derived templates, it does mean that the visual appearance of classes may be misleading - you will get classes that look like the molecular envelope of your protein, even if you pick from entirely random noise (cf Einstein from noise effect). If you saw clear high resolution features I wouldn’t be worried (because your templates are filtered to 20 Å), but given that features are ambiguous I would maybe reconsider the picking approach.
I would always recommend for an initial analysis using some kind of unbiased approach - either manual picking or blob-based picking, which will usually give good results if appropriately tuned. Subsequently you can take your best subset of 1000-3000 particles and train a Topaz model, which will often give improved results with small/heterogeneous particles.
Cheers
Oli
1 Like
Dear Oli,
The Einstein-from-noise is certainly a chilling possibility  The lack of high-resolution features in the reconstruction is indeed a bit worrying considering that I did supply an initial template for particle picking. I’ll try out the blob→Topaz particle picking pipeline and see if I get a different reconstruction. I probably should’ve looked into the upstream steps a little more altogether, now as you point it out (maybe I got a bit too excited after seeing the ab initio results
The lack of high-resolution features in the reconstruction is indeed a bit worrying considering that I did supply an initial template for particle picking. I’ll try out the blob→Topaz particle picking pipeline and see if I get a different reconstruction. I probably should’ve looked into the upstream steps a little more altogether, now as you point it out (maybe I got a bit too excited after seeing the ab initio results  ).
).
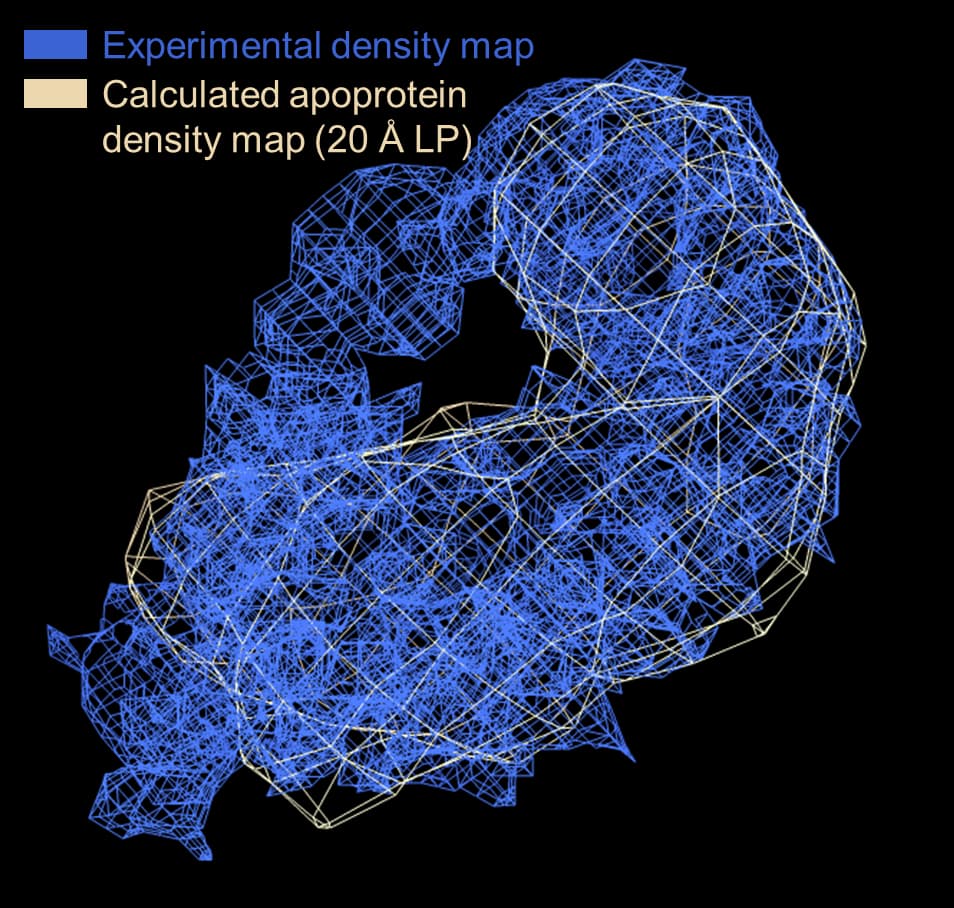
That said, hopefully the current reconstruction still isn’t pure noise since I see a tube of strong (stronger than parts of the apoprotein) density extending beyond the apoprotein envelope (i.e. supplied template) that appears to be the bound RNA ligand. (The particles used for reconstruction are also mostly visible by eye upon manual inspection)
Thanks for your prompt reply and happy holidays!!
Sincerely,
Joonyoung
I have tried out the unbiased particle picking approach as @olibclarke suggested. Specifically, I used blob picker to get ~450K picks from a sample stack of 500 micrographs and went through multiple rounds of 2D classification to select ~50K particles showing protein-like density. Moving onto multi-class ab initio, I was able to get a “good” 3D class resembling the density map I had observed previously in earlier template-based analyses. (At least we can rule out the Einstein-from-noise  )
)
I used a subset of ~3K particles (spread over 100 micrographs) from the “good” 3D class to train a Topaz picker model. The model appeared to be well-trained and generated reasonable particle picks when tested on micrographs outside the training dataset.
Unfortunately, the overall results on 3D reconstruction/refinement were similar as before, with any attempts at increasing resolution beyond 10Å simply leading to noise overfitting. I am starting to suspect that the quality of the micrographs themselves might be the culprit. Comparing against micrographs from a recent Cryo-EM study (Abeywansha et al., 2023) of a similarly-sized & shaped complex, my micrographs look a bit less-defined and featureless. I don’t know if this is due to differences in detector technology (Falcon 4i vs K2), ice thickness, or some other factor. I’m not even sure if the “amorphous” look in my micrographs is necessarily a bad thing (i.e. indicative of data that cannot be refined to high resolution)  .
.
I wonder if anyone with experience with proteins in this size range (50-60 kDa) could enlighten me on whether this resolution problem is more likely an issue with the raw data or my approach at processing the data.
Thanks to @olibclarke as always and happy new year everyone!
Sincerely,
Joonyoung
Example 2D classes from blob picks after few rounds of cleaning
Topaz training data
Example particle picks using the Topaz model (defocus 2.4 μm, 5Å LP-filtered)
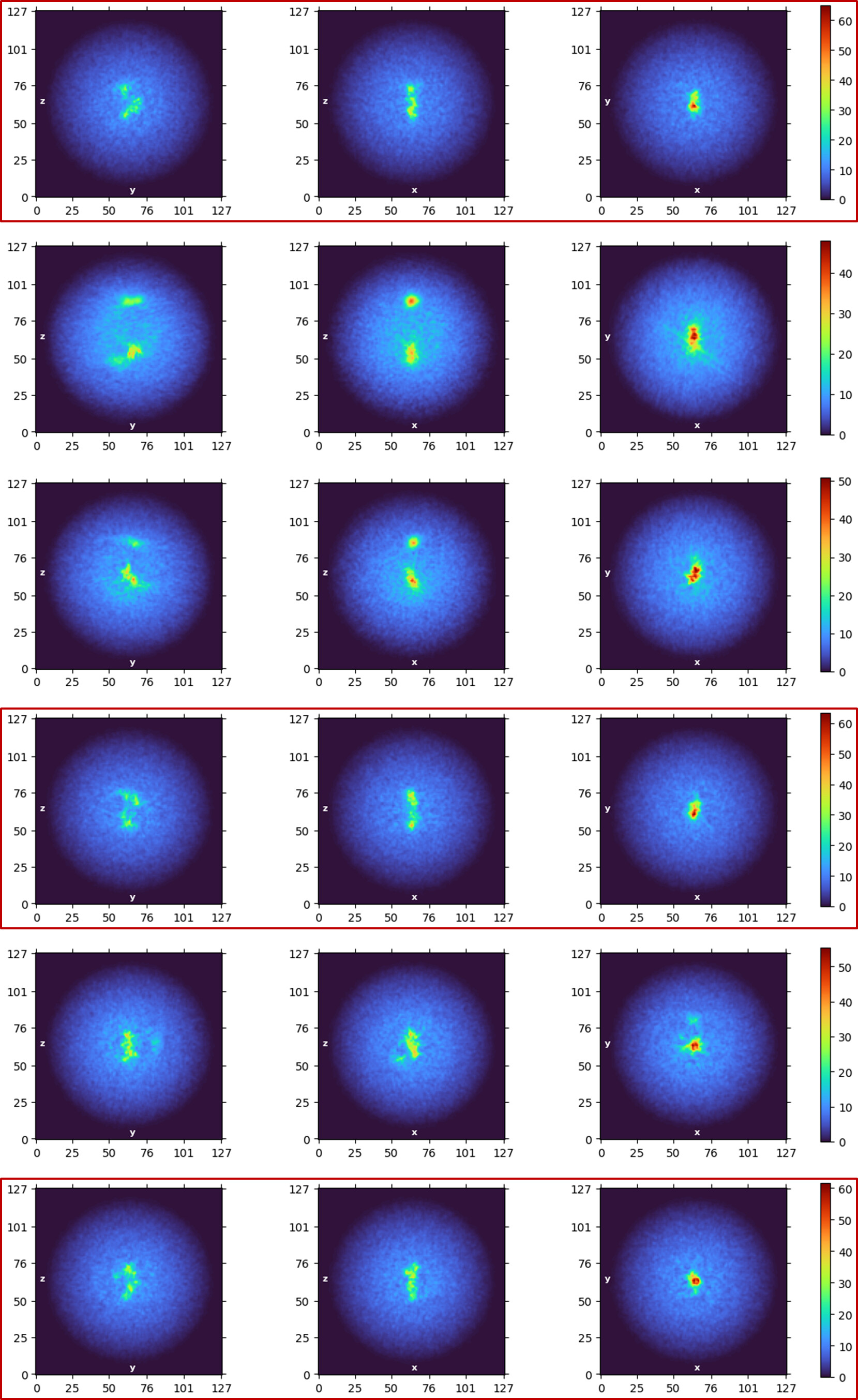
Multi-class ab initio 3D reconstruction from Topaz picks (good classes outlined in red)
Example micrograph & 2D classes from Abeywansha et al., 2023
Hello Joonyoung,
Can you please show me the detail parameters of your 2D classification? Actually, I got a small size protein recently, which is around 67 kDa and form the complex with 15-Mer RNA. The same to your case, I have tried turn off force max over poses/shifts and increase online EM iterations to 40-80, increase batchsize per class to 200-400, lower recenter mask threshold to 0.1. None of them is work. My pixel size is 0.743, I tried box size 256, 288, 320, none of them is work.