I have a C15 map in which I want to start model building. I roughly fitted one monomer in ChimeraX and I tried to copy it to make the 15-mer. This failed at first because the default position for my map when opened in ChimeraX is obviously not the original symmetry axis point (I don’t know why). After some troubleshooting, I found that the even the displayed symmetry axis in ChimeraX is slightly offset and I do not know how to get the correct coordinates. Is there a way to get this information directly from the cryoSPARC output of the map I am using?
Hi Noha - how did you obtain the C15 map? How do you know the symmetry axis is offset?
I would recommend using servalcat to generate the oligomer, e.g.
servalcat util symmodel --model monomer.pdb --chains A --pg C15
Cheers
Oli
I obtained the C15 map by using a C15 symmetry in NU-refinement, after observing the C15 symmetry in the top view of my 2D classes. It is the reported symmetry group for my particle as it has been solved before.
I know it is offset as I can see this in Chimera. First when I tried to copy the monomer I fitted (in coot) in chimera to generate a C15 model, I noticed that the position where the monomers were copied was wrong and not inside the map. I then found the original index point from the map coordinates and moved the map to those coordinates. The problem I faced next was that my monomer had to be moved to the original map coordinates and then copied. I attached a photo to demonstrate. If you look at the map you will notice that it is not in the center of the ring of monomers. In the end I got the C15 model, but when I opened it in coot, I could see that there is a gradual shift with every copied monomer in the model. I tried to run one cycle of rigid body refinement in phenix so that it might compensate for this slight shift, but it didn’t.
I will try to install and run servalcat, but honestly coding is still not easy for me so I am not sure if I will be able to do it. That is why I was trying to get the original map coordinates from cryoSPARC to try to copy the monomers in coot or chimera.

Servalcat is included with CCP-EM - so if you install CCP-EM, you will have servalcat.
It sounds like what you are describing is an inconsistency between Chimera and Coot - that is, you fit in Coot, and when you open in Chimera it doesn’t match the map, is that right? What happens if you fit your monomer in Chimera to begin with?
The ring of monomers you are showing is far away from your map - is this just for illustrative purposes, presumably this is not the C15 model you describe?
I did an initial rough fitting of my monomer in the map using chimera. This “fitting” was done by fragmenting my monomer into domains in PyMoL then doing “fit in map” using chimera until its fit as best as could be at this stage. When I had the whole monomer, I took the map and the monomer model to coot, to improve the fitting by refining the residues one by one. When I was done with that I tried to find a way to just copy the monomer that I exported from coot (didn’t know how to copy in coot) to have a 15-mer. To do this, I tried to use the following command:
sym #model no. C15 copies true
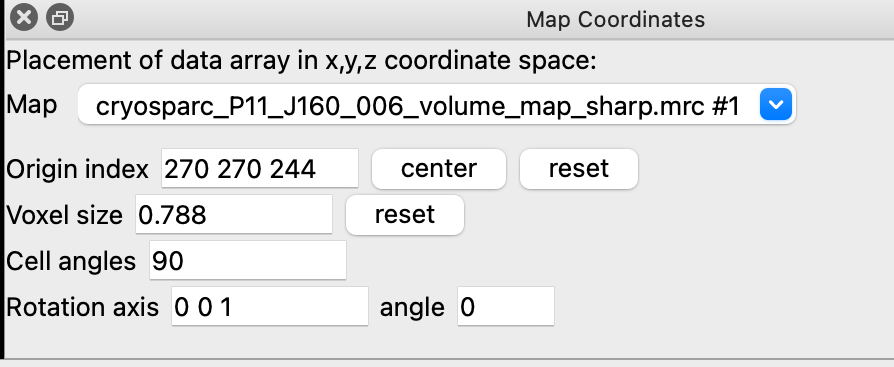
When I did that I ended up with the weird picture I attached (only that the model was in the center of the screen as I haven’t tried to move the map then yet from its coordinates). I got back to the Chimera mailing list to try to understand what I was doing wrong. I learned then that I could use the (menu Tools / Volume Data / Map Coordinates) in chimera to get the original origin grid index (270, 270, 244, I also attached what I get). I then used the following commands:
volume #map no. originIndex 270,270,244 (it moved)
sym #model no. C15 copies true
Here I ended up with the weird picture I attached in the previous message. The monomers still did not fit into the map. That is because my monomer was now in a different position relative to the map after it moved. To solve this problem, I used the following command:
move -2700.788, -2700.788, -244*0.788 model #
Where 0.788 is my voxel size. When I did this, followed by the sym command, I could finally obtain the 15-mer, and it looked perfect. However, to confirm if there was no offset, I was advised to use the following command and see if the map would wobble:
turn z 24 frames 150 model #no.
Which it unfortunately did. In the attached “map coordinates” if you press “center” the origin index becomes “269.5”, which I believe indicates that this is the true X coordinate or Y I am not sure. So the 270 270 244 is indeed offset.
Now if I take the map and the model to coot, they wouldn’t align at all since the original map file is not in the original grid index, unlike the model I just saved. So I had to save the model in the original coordinates and they finally align in coot, all by it with the slight offset.
Now, I am also trying to run a script to copy the one fitted monomer (before all this mess happened) into the map directly in coot, without having to resort to chimera at all…but for this, I need the transformation matrix from the cryoSPARC output, or as far as I understand, the original map coordinates…
When I did that I ended up with the weird picture I attached (only that the model was in the center of the screen as I haven’t tried to move the map then yet from its coordinates). I got back to the Chimera mailing list to try to understand what I was doing wrong. I learned then that I could use the (menu Tools / Volume Data / Map Coordinates) in chimera to get the original origin grid index (270, 270, 244, I also attached what I get). I then used the following commands:
volume #map no. originIndex 270,270,244 (it moved)
sym #model no. C15 copies trueHere I ended up with the weird picture I attached in the previous message. The monomers still did not fit into the map. That is because my monomer was now in a different position relative to the map after it moved. To solve this problem, I used the following command:
Check the CRYST1 line in your PDB. Chimera relies on this being correct for the sym command to work. Chances are it is wrong (inherited from an original crystal structure or model). So alter it to match the dimensions of your map (in Å) and it should work.
Remember your PDB is just a text file - open it and look for the line that starts with CRYST1. This will have the cell dimensions. Alter these to match the map, and delete the lines underneath that start with SCALE, if present. I expect that should fix the issue.
Alternatively (maybe easier), you can specify the map as the coordinate system for the sym command - so instead of sym #model no. C15 copies true, use sym #model no. C15 copies true coordinateSystem #map no..
Also - what is your box size in pixels? It should usually be an even number - if it isn’t, I would recommend redoing your refinement with an even-numbered box size, as odd numbered boxes tend to cause issues for other programs.
1 Like
Hello everyone!
I have a similar issue. Let me explain it.
I made a fitting model with my density map, it had been create on cryosparc. I didn’t have any problem to the fitting process on chimeraX. However, when I finished the refinement on phenix and coot, I wanted to finish the atomic model with the sym option in chimera x, but the result is similar to Noha’s.
did you check the CRYST1 flag as per the previous message in this thread? Or alter the coordinate system to use for sym?
Has anyone solved the problem? I’m running into the same issue and can’t see the CRYST1 flag in the PDB file. Unsure why. My case is very similar to what Noha described, but with a C19 symmetry. Haven’t been able to figure it out. Any idea?
Thanks!
When opening chimera is the symmetry expanded C1 monomer local refine map nicely inside where is should be in C19 map? If not then issue is there, if doesn’t then was the box size changed for local refinement?
With a fit monomer model in the C19 map, have you tried to symmetry multiply in phenix rather then chimera?
Phenix.map symmetry (CryoEM/Map Symmetry) - find symmetry on C19 NU-refine or C19 local refine map; not on C1 local refinement of symmetry expanded.
Phenix.Apply NCS operators (CryoEM/Modification/Apply NCS operators) -applies symmetry monomer model + previous map symmetry
Composite map? Need to check potential symmetry clashes.
Hi all, has anyone found a way around this issue?
I’m in the same boat as @Noha_Elshamy with several models, most of which are in C6. I have a full monomer of each protein built for each model, but cannot seem to get them to symmetrize correctly with respect to their maps. I built the monomers in Coot.
Things I’ve tried:
- Manually attempting to symmetrize in ChimeraX by determining the centre of symmetry for the map and applying it to the model alongside its symmetry parameters (and using the coordinateSystem command)
- Shifting the model based on voxel size (ie. what Noha had tried based on previous advice; generated the same result)
- Altering the CRYTS1 line in my PDB and deleting SCALE parameters (CRYST1 was correct to begin with and pixel size is even)
- Using Phenix to determine NCS operators given the C6 local refinement maps (generated a blank text file despite detecting correct symmetry in the log, unsure why)
- Using the “dock in model” and “map to model” options in Phenix (did not finish running even after 3 days)
In ChimeraX, the monomers fit perfectly into their maps. I can get them to “symmetrize” into what looks like a full build based on the parameters I’ve already determined (including helical parameters for some) but the maps and models cannot seem to agree on a centre or orientation – almost like the monomers are being duplicated backwards (for lack of a better word) in some cases.
Aside from manually inserting each monomer copy into the builds, I’m at a loss for options. Wondering if anyone has had some luck with other programs as well.
For ChimeraX was the center cmd also used in with the coordinatesystem? Turn/sym need the center of the coordinate system set for the turning point.
sym #1 C6 coordinatesystem #2 center 240,240,240
#1 LR model, #2 C6 NU map, the center x,y,z in angstrom so box pixel width * pixel size / 2 for center (here 480 box size with 1A pixel size)
For C6 need to tell it the center as z-axis, which can randomly get it work if open a C6 map and orients on the Z-axis initially, or use the view orient cmd with just C6 map open as same thing.
For phenix map symmetry, something to try would be on another C6 map (own or emdb) to get what the file output looks like and put in your boxsize and center for your maps and use that as input for aNCS job