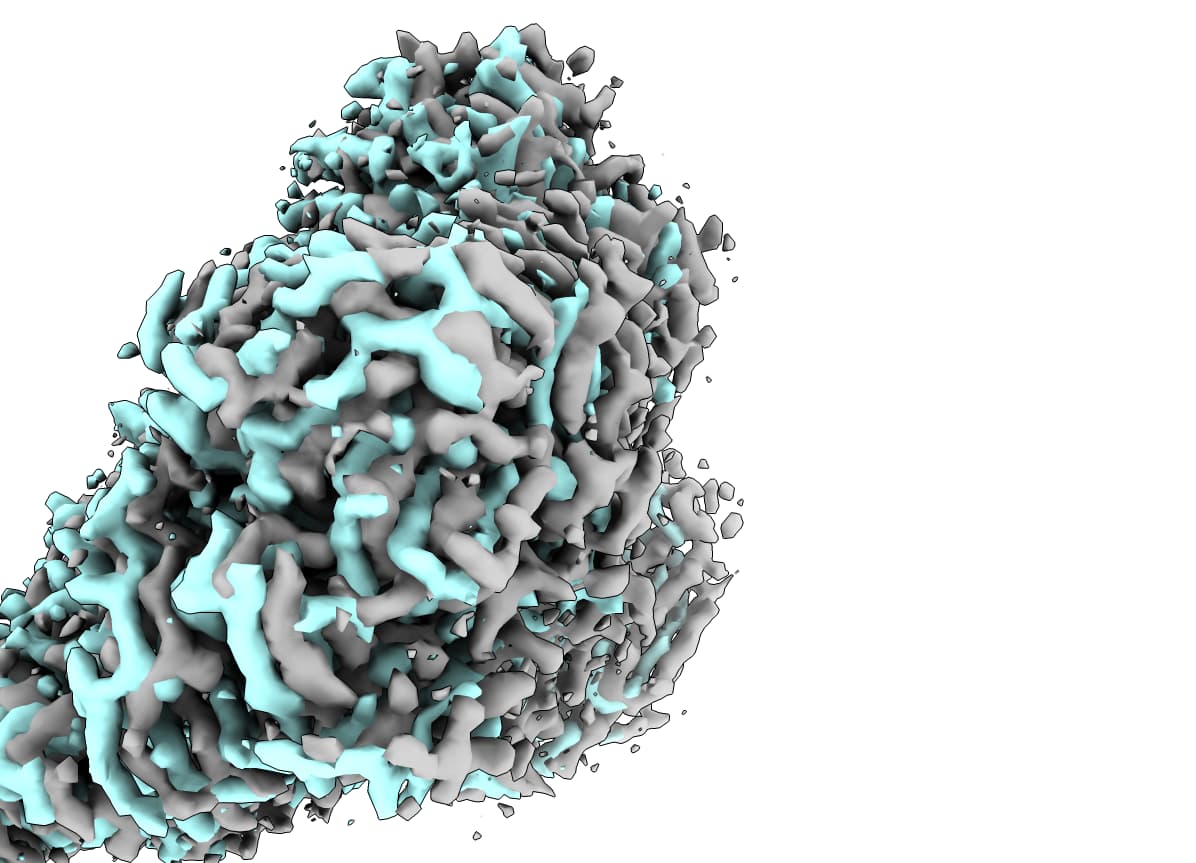
I have a few datasets that will have unique depositions, and was hoping to deposit them in the correct alignment/register. I can perfectly align the maps in ChimeraX, but ‘Align 3D Maps’ in CryoSPARC doesn’t quite work.
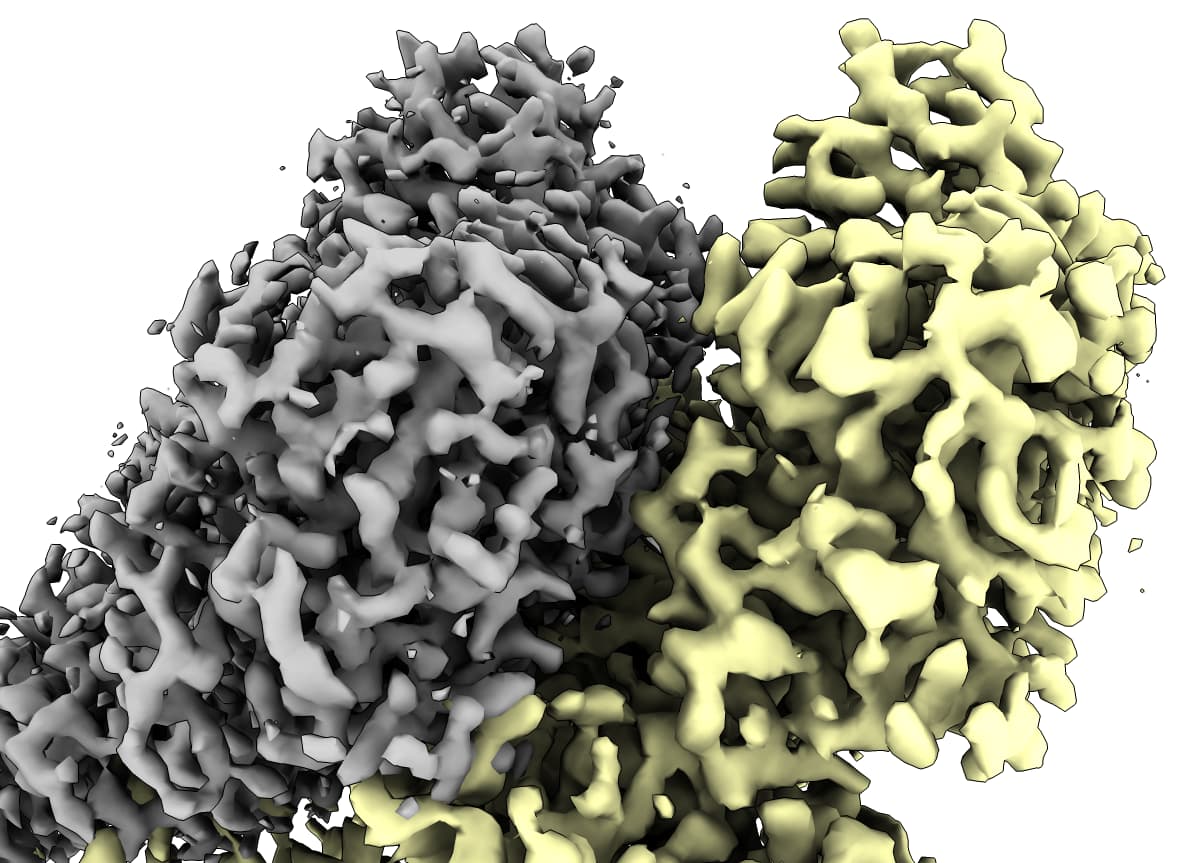
In grey (dataset1) is the reference map, cyan (dataset2) is a A3DM map that is just slightly offset, yellow (dataset3) is a correctly oriented A3DM map but translated quite far from the grey map. I’ve tried to connect the output of one round of A3DM to a subsequent round, but the output is exactly the same as the first, so it seems to me like the A3DM results are very close but get “stuck” in some minima.
Are you using a mask for the alignment? And have you tried playing with the resolution for alignment? Definitely Align3D can be a bit finicky sometimes…
I have tried A3DM with/without masks, and various low-pass filters (3 - 12 A), and the result is the exact same each time (at least for the cyan → grey alignment example).
Are you referring to the Phenix ‘Superpose maps’ job? I found this thread (superpose CryoEM map onto an Xray map? - phenixbb - phenix-online.org) that describes first converting your MRC maps to MTZ maps for the job, though Pavel mentions that there are still artifacts that can propagate if the maps are not on the same grid. I don’t see where it accepts a matrix file, and I tried matrixcopy with ChimeraX, but that command does not seem available anymore – is there an updated one you can share? I found matchmaker in ChimeraX has the ‘Log tranformation matrix’ checkbox in the Fitting tab, is that sufficient?
I am not sure if there is an implementation of matrixcopy in ChimeraX - it is present in Chimera, which still works fine
I wasn’t referring to superpose maps in Phenix, just to resampling maps in Chimera. This would be at the end of your workflow - after refinement. I wouldn’t redo refinement after aligning, as this may subtly mess up the alignment (assuming you are starting refinement at a relatively low resolution).
I have been reading through Kleywegt et al., 2024 (DOI: 10.1107/S2052252524001246), and was hoping to satisfy the recommendations list. So, by resampling maps in Chimera onto a reference map, how can I also deposit the relevant masks/half-maps/particle stacks?
You can apply the same transformation (using matrixcopy) to the half-maps/masks, and resample on the same grid. Or just live with them unaligned.
Those recommendations are good general guidelines; but there are always going to be exceptions, which is fine if they are well reasoned & rationalized, just explain what you have done and why (IMHO).
For anyone that may find this useful, I am providing the instructions for doing the matrixcopy Dr. Clarke referred to (I couldn’t find very clear instructions online).
In Chimera (not ChimeraX) load: #0: reference map (to be aligned to) #1: map to be moved #2: reference PDB (of map #0) #3: PDB for map to be moved (of map #1)
I recommend that you have previously saved the reference PDB as the same model fit individually fit into both the #0 and #1 maps so that the rotation/translation will be consistent.
run Matchmaker (Tools → Structure Comparison) of #3 aligned to #2
open Reply Log (Tools → Utilities → Reply Log)
Follow up to @dbuckley, ChimeraX has this built into the gui matchmaker, bring map with model.
Tools → Structure Analysis → Matchmaker
Reference structure: (your stable model)
Structure to match: (your mobile model)
Fitting tab (half way down the gui)
If one model being matched, also move these models along with it:
Choose map that mobile model fits in
Apply
volume resample #2 (mobile map) ongrid #1 (stable map)
cli
matchmaker #4 to #3 bring #2
volume resample #2 (mobile map) ongrid #1 (stable map)
help matchmaker
log will give the command and you can repeat in cli with specific regions of model for matchmaker to align if needed
Yes, agreed, I would not recommend resampling for deposited maps. If you want everything to be easy to overlay, resampling references and re-refining is the only way I’d suggest.
I wanted to do this, but I’ve found that using the resampled maps in subsequent refinements results in very odd behavior. I can attach some examples if needed. I have 6 datasets obtained from different scopes with varied pixel sizes, so I’m assuming it’s related to that. Thus, I was hoping the Align 3D Maps job would remedy this, but that’s where I got the suboptimal alignments.
Definitely curious, as the reference should be lowpass filtered enough that it shouldn’t do anything strange.
That said, I do remember being confused with something in an earlier version of CryoSPARC (3.2, I think… I forget exactly) where initial iterations of refinement looked a bit weird when using a map I’d previously generated in RELION as a reference, but it recovered around iteration 4 and gave a good final reconstruction. At the time, I put it down to not having enough CryoSPARC experience…
I’ve tried a number of combinations to try to get this to work. For two of my datasets, the reference dataset (#1) has a pixel size of 0.938 A and the dataset I’d like to move (#2) has a pixel size of 0.928 A (collected on different microscopes). So, I’ve tried uploading the map of #2 into CryoSPARC after aligning and resampling to #1 in Chimera and rerunning my latest NU-refinement with this new reference #2 volume. That resulted in the offset that I showed an image of at the top of this thread, where the grey map is the reference and the cyan is the map after NU-refine (in addition to a worsened cFAR score for some reason). This exact result happens with running the Align 3D Maps job as I previously mentioned.
I tried importing the aligned #2 volume and overriding the pixel size at that step, but it results in the same offset. My only idea is that I need to figure out an override pixel size that compensates for the offset, but I’m not sure how to go about that. Is there some way that the Align 3D Maps job could incorporate maps of different voxel sizes?
Hopefully I’m describing this well, please let me know if I can provide any additional info.
CryoSPARC can resample on the fly, so try feeding map #2 to dataset 1 as a reference directly (without aligning/resampling). If centred and aligned to desired symmetry, turn off symmetry alignment in homogeneous/NU refinement - a dataset I was processing last night is nicely centred in C1, but always ends up wildly off-centre on the Z-axis if I ask CryoSPARC to align to C2…
Ahh the different pixel sizes makes it a bit more complex. Are you using the same box dimensions (512 box size for both .928 and .938 datasets)? Have the two datasets been pixel calibrated (ChimeraX: Volume → map coordinates → voxel size, adjust pixel size and volume → fit in map to get correlation with smaller and smaller increments for voxel size), this is only if expecting the maps to not be different in area of calibration, can do with model fit also.
Have you tried moving map #1 to location of map #2 then resampling #1 moved on a copy of map #1 in original location. That would ignore the pixel size difference, which is small so minor shift in particles. Rerun refinement using that new model. Two datasets 1% off pixel size so moving just a few pixels off center if box size same size for different pixel size. Can also try in cyrosparc 3D align map #1 as reference, then align map “#1 moved and resampled on #1 original” along with particles. 3Dalign works rather well when giving it the same map but moved, homogenous reconstruction and see if they align now.
Just some info that might be helpful. If using ChimeraX switch the background to black (home/background/black or cli: set bgcolor black). Then turn on the box (Map/outline box or cli volume #* showoutlinebox true). If your boxes are the same dimensions 512 pixels, but different pixel size .928 vs .938 the boxes should look different sizes. Resampling grabs the new box (both the pixel size and dimensions), anything outside of box being resampled on is lost, having different pixel sizes should make resampling worse is resolution loss.
Side note: as the step size of map affects the voxel size so if resampling on a map that step 2 would get 1.856 pixel size, as @rbs_sci mentions cryosparc would grab that map and see the different in size (box size?) and resize for use, this is useful to make masks that don’t take up as much storage, resample onto original map that is currently set to stepsize 2, the 512 box size-> 256 so storage size 1/8th. This however affects the volume tools job as your pixel size now is different so need to adjust dilation/softness accordingly as those are based on number of pixels.
thanks everyone for your suggestions, just thought I’d give a final update.
@rbs_sci, I was able to use my map #2 (from dataset 2) as a direct volume reference for NU refinement for dataset 1. Unfortunately, that didn’t quite work when I tried to use map #2 as a reference in NU refinement for dataset 3 (these are all Fab-bound structures, so datasets 1/2 have highly similar epitope/paratope interactions, while dataset 3 is quite different).
I found the best success in going back to using the Align 3D tool. Since the antigen is the same for all my datasets, I split that portion of the volume away from the Fab for dataset 2
Then import this new map into CryoSPARC with ‘Type of volume being imported’ = map
This allowed me to update all the particle alignments in A3DM and run a final NU-refinement job (free of any resampling artifacts we discussed earlier).
And thanks to @T_Bird, I did find there was a slight disagreement between some of the boxsizes for the different datasets, so I’ve standardized all that as well.
The maps are not as perfectly aligned as you can get within ChimeraX, but at the very least they are not wildly out of alignment. I am probably heavily overestimating how much my maps will be looked at, but I was hoping to save people trouble in deciphering how to align all these structures!